Search

The MEGAscript RNAi Kit is a system for the preparation of double-stranded RNA (dsRNA), free of protein and other contaminating molecules, for use in RNA interference (RNAi) or other experiments. This kit is designed for the preparation of dsRNAs larger than ~200 bp. For preparation of small interfering dsRNA, known as siRNA (~20 bp long), we recommend the Ambion Silencer siRNA Construction Kit, or Custom siRNA Synthesis Service.
The procedure begins with a high yield transcription reaction based on the Ambion MEGAscript technology, to synthesize two complementary RNA transcripts from template(s) supplied by the user. The RNA strands are hybridized either during or after the transcription reaction to form dsRNA. Next, DNA and any single-stranded RNA (ssRNA) are removed with a nuclease digestion. Finally, the dsRNA is purified with a solid-phase adsorption system to remove protein as well as monoand oligonucleotides. The dsRNA produced can be introduced into the organism of interest by a variety of means, including injection, electroporation, or chemically-mediated transfection, depending on the organism being studied.
RNAi, the phenomenon by which long dsRNAs specifically suppress expression of a target gene, was originally discovered in worms (Fire 1998), but this phenomenon has now been found in a large number of organisms, including flies (Misquitta and Paterson 1999), trypanosomes (Ngo 1998), planaria (Sánchez-Alvarado and Newmark 1999), hydra (Lohmann 1999), and zebrafish (Wargelius 1999). The RNAi mechanism is currently being investigated, but it appears to work through smaller dsRNA intermediates. The parent dsRNA is broken down into these smaller fragments in vivo, and this siRNA directs a post-transcriptional breakdown of the targeted mRNA (Zamore 2000). An unusual feature of this process is that it works non-stoichiometrically and can spread between cells (Fire 1998). RNAi is a powerful method to investigate gene function through suppression of gene expression.
The procedure begins with a high yield transcription reaction based on the Ambion MEGAscript technology, to synthesize two complementary RNA transcripts from template(s) supplied by the user. The RNA strands are hybridized either during or after the transcription reaction to form dsRNA. Next, DNA and any single-stranded RNA (ssRNA) are removed with a nuclease digestion. Finally, the dsRNA is purified with a solid-phase adsorption system to remove protein as well as monoand oligonucleotides. The dsRNA produced can be introduced into the organism of interest by a variety of means, including injection, electroporation, or chemically-mediated transfection, depending on the organism being studied.
RNAi, the phenomenon by which long dsRNAs specifically suppress expression of a target gene, was originally discovered in worms (Fire 1998), but this phenomenon has now been found in a large number of organisms, including flies (Misquitta and Paterson 1999), trypanosomes (Ngo 1998), planaria (Sánchez-Alvarado and Newmark 1999), hydra (Lohmann 1999), and zebrafish (Wargelius 1999). The RNAi mechanism is currently being investigated, but it appears to work through smaller dsRNA intermediates. The parent dsRNA is broken down into these smaller fragments in vivo, and this siRNA directs a post-transcriptional breakdown of the targeted mRNA (Zamore 2000). An unusual feature of this process is that it works non-stoichiometrically and can spread between cells (Fire 1998). RNAi is a powerful method to investigate gene function through suppression of gene expression.
The kit contains the components listed in the following tables to synthesize
20 dsRNAs.
* Store Nuclease-free Water at –20°C, 4°C or room temp
20 dsRNAs.
| Amount | Component | Storage |
|---|---|---|
| 10 mL | Nuclease-free Water | any temp* |
| 20 | Filter Cartridges | room temp |
| 40 | Collection Tubes | room temp |
| 40 μL | T7 Enzyme Mix T7 RNA polymerase, RNase Inhibitor Protein, and other components in buffered 50% glycerol | –20°C |
| 40 μL | 10X T7 Reaction Buffer | –20°C |
| 40 μL | ATP Solution (75 mM) | –20°C |
| 40 μL | CTP Solution (75 mM) | –20°C |
| 40 μL | GTP Solution (75 mM) | –20°C |
| 40 μL | UTP Solution (75 mM) | –20°C |
| 1 mL | 10X Binding Buffer | –20°C |
| 4 mL | Elution Solution | –20°C |
| 40 μL | RNase | –20°C |
| 45 μL | DNase I | –20°C |
| 100 μL | 10X Digestion Buffer | –20°C |
| 12 mL | 2X Wash Solution Add 12 mL 100% ethanol before use | –20°C |
| 20 μL | Control Template (500 ng/μL) | –20°C |
* Store Nuclease-free Water at –20°C, 4°C or room temp
Gene-specific template(s)
A transcription template(s) is needed with T7 RNA polymerase promoters positioned to transcribe sense and antisense RNA corresponding to the target RNA.
For dsRNA purification
- 100% ethanol: ACS grade or better
- Equipment to draw solutions through the Filter Cartridges: Use either a microcentrifuge capable of at least 8,000 X g, or a vacuum manifold with sterile 5 mL syringe barrels mounted to support the Filter Cartridges.
To assess the reaction products
Reagents and equipment for agarose gel electrophoresis
Spectrophotometer
A. Choosing the dsRNA Sequence
RNAi experiments are typically done with dsRNA 400 bp and larger; 200 bp is the minimum size of dsRNA recommended for RNAi. Typically templates for transcription of dsRNA for use in RNAi correspond to most or all of the target message sequence.
B. Strategies for Transcription of dsRNA
RNAi experiments requiredouble-stranded RNA (dsRNA). Since the T7 RNA polymerase used in this kit synthesizes single-stranded RNA (ssRNA), use one of the following strategies to produce dsRNA:
- Prepare one DNA template with opposing T7 promoters at the 5' ends of each strand, and use it in a single transcription reaction to synthesize dsRNA without a separate annealing step.
- Use two DNA templates that are identical except that a single T7 promoter sits at opposite ends of the region to be transcribed. With this strategy, the templates can both be added to a single reaction. Although both templates should be transcribed at the same rate, if they are not, the final dsRNA yield will be dictated by the template with the lower transcription efficiency. Alternatively, the two templates can be transcribed in separate reactions to make complementary RNA molecules, which are then mixed and annealed. For the annealing step, the two transcripts can be mixed in precisely equimolar amounts. Note, however, that if the templates are transcribed in separate transcription reactions, this kit contains enough reagents to produce only 10 different dsRNAs.
Figure 1. T7 Polymerase Promoter: Minimal Sequence Requirement
+1
5'-TAATACGACTCACTATAGGGAGA-3'
|________________________|
The +1 base (in bold) is the first base incorporated into RNA. The underline shows the minimum promoter sequence needed for efficient transcription.
C. PCR Templates
Amplification strategies to add T7 promoter sequences to DNA
T7 promoter sequences can be added to DNA using PCR to generate templates that can be directly added to the MEGAscript RNAi Kit transcription reaction. Begin by synthesizing PCR primers with the T7 promoter sequence appended to the 5' end of the primer. The T7 promoter-containing PCR primers (sense and antisense) can either be used in separate PCRs, or in a single PCR to generate transcription template for both strands of the dsRNA. The two strategies for adding T7 promoter to DNA are shown below:
Figure 1 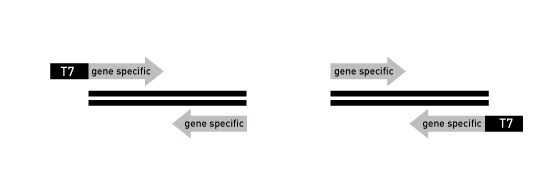
|
|
- Calculate the annealing temperatures of the entire PCR primer (with the T7 promoter site) and the gene specific portion of the PCR primer separately.
- Since the first cycles of PCR use only the 3' half of the PCR primer(s), the gene-specific part, the annealing temperature for the first 5 PCR cycles should be ~5°C higher than the calculated Tm for the gene-specific region of the primer. We have found that using the calculated annealing temperature for the initial cycles often results in synthesis of spurious PCR products.
- Once some PCR product is made, subsequent primer annealing events (cycle 6 and thereafter) use the entire primer site; therefore use the calculated Tm for the entire PCR primer plus ~5°C for subsequent cycles.
- We recommend using primers at 100 nM in the PCR mix. Higher concentrations may result in synthesis of primer dimers.
Check PCR products on a gel before using them in this procedure
PCR products should be examined on an agarose gel prior to in vitro transcription to estimate concentration and to verify that the products are unique and of the expected size.
D. Plasmid Templates
Cloning strategy
When using plasmid templates in the MEGAscript RNAi Kit, it is best to make two separate clones with the same target region in both orientations.
Figure 2. Cloning in plasmids 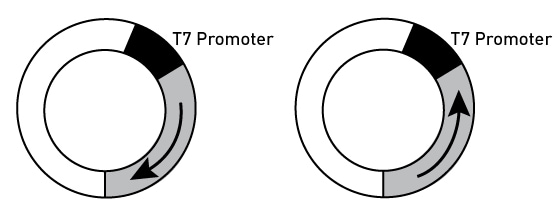
Note: Although some dsRNA templates have been cloned in plasmids containing opposing T7 promoters (on either side of the linker region Kamath 2001, Timmons and Fire 1998, Morris 2001), these constructs will result in synthesis of dsRNA with significant stretches of RNA corresponding to the polylinker region of the plasmid.
PCR products can be cloned into plasmid vectors using any of the following strategies:
- Amplify the target by PCR and ligate the product into a PCR vector with a T7 promoter. Identify plasmids with the insert in both orientations with regard to the T7 promoter.
- Or, include the T7 promoter sequence at the 5' end of one or both of the PCR primers, then perform PCR to incorporate them into the fragment. Finally, ligate the PCR product into a PCR cloning vector (one that does not include a T7 promoter).
- Or, include both a T7 promoter and a restriction site at the 5' end of one or both PCR primers to incorporate them into the fragment during PCR. Ligate the PCR product into a cloning vector using the added restriction sites.
Plasmid linearization
Plasmid templates must be linearized downstream of the insert to create a transcription termination site—the RNA polymerase will literally fall off the end of the DNA molecule. Linearize each template, then examine the DNA on a gel to confirm that cleavage is complete. Since initiation of transcription is the rate limiting step of in vitro transcription reactions, even a small amount of circular plasmid in a template prep will generate a large proportion of transcript, wasting much of the synthetic capacity of the reaction.
Note: We routinely use all types of restriction enzymes. However, there has been one report of low level transcription from the inappropriate template strand in plasmids cut with restriction enzymes leaving 3' overhanging ends (produced by Kpn I, Pst I, etc.; Schenborn and Mierindorf, 1985).
Figure 3 Linearized plasmids
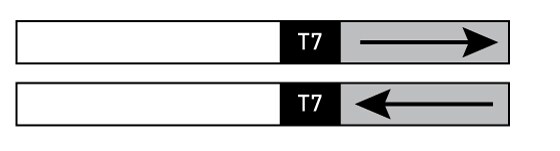
After linearization
Terminate the restriction digest by adding each of the following:
- 1/20 volume 0.5 M EDTA
- 1/10 volume of either 3 M NaOAc or 5 M NH4OAc
- 2 volumes of ethanol
Mix well and chill at –20°C for at least 15 min. Then pellet the DNA for 15 min in a microcentrifuge at top speed. Remove the supernatant, respin the tube for a few seconds, and remove the residual fluid with a very fine-tipped pipet. Resuspend in TE (10 mM Tris-HCl pH 8, 1 mM EDTA) at a concentration of 0.5–1 μg/μL.
Plasmid DNA purity
DNA should be relatively free of contaminating proteins and RNA. The greatest yields of dsRNA will be obtained with very clean template preparations. Most commercially available plasmid preparation systems yield DNA that works well in the MEGAscript RNAi Kit. Alternatively, a DNA miniprep procedure that generally yields high quality template is presented in section V.D.
Note that DNA from some miniprep procedures may be contaminated with residual RNase A. Also, restriction enzymes occasionally introduce RNase or other inhibitors of transcription. When transcription from a template is suboptimal, it is often helpful to treat the template DNA with proteinase K before performing the transcription reaction.
A. Before Using the Kit for the First Time
Prepare the Wash Solution
1. Add 12 mL ACS grade 100% ethanol to the bottle labeled 2X Wash Solution.
2. Mix well, and store at room temperature.
We suggest crossing out the 2X from the bottle label after adding the ethanol. In these instructions this reagent will be called Wash Solution once the ethanol is added.
B. Transcription Reaction Assembly
1. Thaw the frozen reagents at room temp then place them in ice
Remove the T7 Enzyme Mix from the freezer and place it directly in ice; it is stored in glycerol and will not freeze at –20°C. Vortex the 10X T7 Reaction Buffer and the 4 ribonucleotide solutions (ATP, CTP, GTP, and UTP) until they are completely in solution. Once they are thawed, store the ribonucleotides (ATP, CTP, GTP, and UTP) on ice, but keep the 10X Reaction Buffer at room temperature. Microcentrifuge all reagents briefly before opening to prevent loss and/or contamination of any material on the rim of the tube.
2. Assemble transcription reaction at room temperature
Assemble the transcription reaction at room temperature in the order shown below. The following amounts are for a single 20 μL transcription reaction. Reactions may be scaled up or down if desired.
Important: The following reaction setup is recommended when the RNA produced will be ≥400 nt in length. For transcripts shorter than this, see section V.C. Optimizing Yield of Short Transcripts for modified reaction setup suggestions.
| Amount | Component |
| to 20 μL | Nuclease-free Water |
| 1–2 μg | Linear template DNA Use either 1 μg of a template with opposing T7 promoters flanking the transcription region, or use a mixture of 1 μg of each template when the T7 promoter template is on separate molecules |
| 2 μL | 2 μL 10X T7 Reaction Buffer |
| 2 μL | ATP Solution |
| 2 μL | CTP Solution |
| 2 μL | GTP Solution |
| 2 μL | UTP Solution |
| 2 μL | T7 Enzyme Mix |
3. Mix thoroughly
Gently flick the tube or pipette the mixture up and down, then briefly microcentrifuge to collect the reaction mixture at the bottom of the tube.
4. Incubate at 37°C for 2–4 hr
The first time a new template is transcribed, the recommended incubation time is 2–4 hr. The optimal incubation time for a given template varies depending on its size and transcriptional efficiency. For transcripts <400 nt, a longer incubation time (up to ~16 hr) may be advantageous, since more transcription initiation events are required to synthesize a given mass amount of short RNA, compared to transcription of longer templates. Instead of doing overnight incubations, we suggest freezing the reaction overnight and then thawing and resuming the reaction the next day. Reactions are stable for several days when frozen at –20°C; however, storage will result in gradual loss of enzyme activity. To determine the optimum incubation time for maximum yield with a given template, a time-course experiment can be done. To do this, set up a transcription reaction, and remove aliquots of the reaction at various intervals (for example after 2 hr, 4 hr, 6 hr, and overnight incubation). Assess results by looking for a decrease in absorbance at 260 nm as the free nucleotides are incorporated into RNA, or by running 0.5 μL samples of the RNA on a gel stained with ethidium bromide
C. Annealing RNA to Maximize Duplex Yield
Include this annealing step for the following types of reactions:
- All >800 nt dsRNA synthesis reactions
- ≤800 nt dsRNA synthesis reactions when the two strands were synthesized from separate transcription templates (in the same or in separate transcription reactions).
Annealing the complementary RNA is often unnecessary for transcripts ≤800 nt made from a single template with opposing T7 promoters because RNA products in this size range will typically hybridize during the transcription reaction. With transcripts >800 nt, however, at least a portion of the transcripts form aggregates (presumably branched structures) rather than the dsRNA.
1. Mix the transcription reactions containing complementary RNA
- If sense and antisense RNA were synthesized in separate transcription reactions, add the entire contents of one of the reactions to the other. If desired, reserve a 0.5 μL aliquot of each template before mixing for gel analysis.
- If sense and antisense RNA was synthesized in a single transcription reaction, both strands of RNA will already be in a single tube; proceed to step 2
2. Incubate at 75°C for 5 min, then cool to room temperature
Incubate at 75°C for 5 min then leave the mixture on the bench to cool to room temperature. The RNA will anneal as it cools, forming dsRNA. Do not put the reaction on ice to cool.
3. Check 1/400th of the dsRNA on an agarose gel
Run 1/400th of the dsRNA on a 1% agarose gel (nondenaturing) to examine the integrity and efficiency of duplex formation.
- 1/400th of a 20 μL dsRNA solution is 5 μL of a 1:100 dilution.
- Dilute the gel samples in TE (10 mM Tris, 1 mM EDTA) or in gel loading buffer.
(Instructions for running a gel are in section V.B.) The dsRNA will migrate slightly slower than DNA markers of the same length.
D. Nuclease Digestion to Remove DNA and ssRNA
This DNase/RNase treatment digests template DNA and any ssRNA that did not anneal. RNase will not degrade dsRNA when using the reaction conditions specified below.
1. Assemble RNase digestion reaction on ice
The amounts shown are for a 20 μL transcription reaction; scale up if your transcription reaction was larger.
| Amount | Component |
|---|---|
| 20 μL | dsRNA |
| 21 μL | Nuclease-free Water |
| 5 μL | 10X Digestion Buffer |
| 2 μL | DNase I |
| 2 μL | RNase |
The ssRNA will be digested after 15 min but allow the incubation to proceed for 1 hr to completely digest the DNA template. Do not continue this incubation longer than 2 hr.
E. Purification of dsRNA
This purification removes proteins, free nucleotides, and nucleic acid degradation products from the dsRNA.
Note: For the quickest dsRNA purification, preheat the Elution Solution to ~95°C before starting the purification procedure.
1. Assemble the dsRNA binding mix
Assemble the dsRNA binding mix by adding 10X Binding Buffer, water, and 100% ethanol to the dsRNA according to the table below.
| Amount | Component |
|---|---|
| 50 μL | dsRNA (from step D.2 above) |
| 50 μL | 10X Binding Buffer |
| 150 μL | Nuclease-free Water |
| 250 μL | 100% Ethanol |
Gently mix the reaction by pipetting up and down.
2. Apply binding mix to the Filter Cartridge, and draw it through
Pipet the entire 500 μL dsRNA binding mix onto the filter in the Filter Cartridge, and draw it through by centrifugation or with a vacuum manifold.
Centrifuge users:
- a. For each dsRNA sample, place a Filter Cartridge in a Collection Tube. Use the Collection Tubes supplied with the kit.
- b. Pipet the entire 500 μL dsRNA mixture onto the filter in the Filter Cartridge. Centrifuge at maximum speed for 2 min.
- c. Discard the flow-through and replace the Filter Cartridge in the Collection Tube.
Vacuum manifold users:
- a. For each dsRNA sample, place a 5 mL syringe barrel on the vacuum manifold, load it with a Filter Cartridge, and turn on the vacuum.
- b. Pipet the entire 500 μL dsRNA mixture onto the filter in the Filter Cartridge. The vacuum will draw the lysate through the filter.
3. Wash the Filter Cartridge with 2 X 500 μL Wash Solution
IMPORTANT
Verify that 12 mL of 100% ethanol was added to the 2X Wash Solution.
- a. Pipet 500 μL of Wash Solution onto the filter in the Filter Cartridge. Draw the wash solution through the filter as in the previous step.
- b. Repeat with a second 500 μL of Wash Solution.
- c. After discarding the Wash Solution, continue centrifugation, or leave on the vacuum manifold for ~10–30 sec to remove the last traces of liquid.
4. Recover the dsRNA 2 X 50–100 μL Elution Solution
- a. The Elution Solution provided with the kit is 10 mM Tris-HCl pH 7, 1 mM EDTA. It is compatible with dsRNA injection, or 2X Injection Buffer can be added to the purified dsRNA for a final concentration of 1X Injection Buffer. Alternatively, the dsRNA can be eluted into any sterile low salt solution (≤30 mM), e.g. 5 mM KCl, 0.1 mM sodium phosphate buffer as used by Rubin and Spradling (1982).Transfer the Filter Cartridge to a fresh Collection Tube.
- b. Apply 50–100 μL (hot) Elution Solution to the filter in the Filter Cartridge.
Apply preheated (≥95°C) Elution Solution to the filter, or
Apply room temperature Elution Solution, close the tube lid over the Filter Cartridge, and incubate in a heat block set to 65°C or warmer for 2 min. - c. Centrifuge for 2 min at maximum speed.
- d. Repeat steps b–c with a second 50–100 μL aliquot of Elution Solution collecting the RNA into the same Collection Tube. Most of the RNA will be eluted in the first elution. The second elution is included to recover any remaining RNA.
5. Quantitate and store the dsRNA
Quantitate the reaction product by measuring its absorbance at 260 nm and calculating the concentration The dsRNA is stable when stored at –20°C in Elution Solution.
6. Check 1/400th of the purified dsRNA on an agarose gel
Run 1/400th of the dsRNA on a 1% agarose gel (nondenaturing) to examine the integrity and efficiency of duplex formation.
- 1/400th of 100 μL elution volume is 2.5 μL of a 1:10 dilution
- 1/400th of 200 μL elution volume is 5 μL of a 1:10 dilution
- Dilute the gel samples in TE (10 mM Tris, 1 mM EDTA) or in gel loading buffer
The dsRNA will migrate slightly slower than DNA markers of the same length.
A. Use of the Control Template
The Control Template is a linear dsDNA fragment with opposing T7 promoters. It yields a 500 bp dsRNA product.
Positive control reaction instructions
- Use 2 μL (1 μg) of Control Template in standard MEGAscript RNAi Kit reactions
- Incubate the transcription reaction for 2 hr.
- Skip section III.C. Annealing RNA to Maximize Duplex Yield as the sense and antisense RNA strands will hybridize during the transcription reaction.
- Follow the procedure as described in sections III.D and III.E starting on to purify the dsRNA.
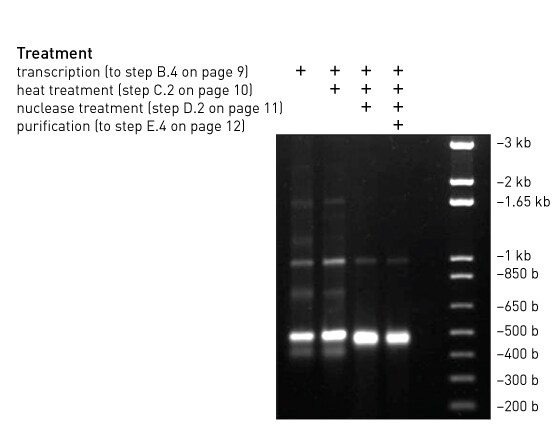
- (Optional) Run 1–2 μg of the positive control reaction product on at 1% agarose gel to verify that the dsRNA is 500 bp.
Expected yield from the control reaction
The yield of RNA from the positive control reaction should be 50–80 μg of a 500 bp dsRNA.
What to do if the positive control reaction doesn’t work as expected
If the yield of RNA from the control reaction is low, something is probably wrong with the procedure, the kit, or the quantitation.
- Double check the RNA quantitation
- When assessing yield by UV spectrophotometry, be sure to use TE (10 mM Tris-HCl, 1 mM EDTA) to blank the spectrophotometer and dilute the RNA.
- To confirm that the quantitation is correct, verify the yield by an independent method. For example if UV spectrophotometry was used to assess yield, try also running an aliquot of the reaction on an agarose gel and comparing its intensity to a sample of known concentration.<
- Consider repeating the positive control reaction. If the yield is indeed low by two different measurements, there may be a technical problem with the way the kit is being used. You may want to consider repeating the positive control reaction. If you are certain that the procedure was carried out correctly, and the control reaction does not give the expected results, contact technical support
B. Low Yield
The amount of RNA synthesized in a standard 20 μL transcription reaction should be 50 μg or more; however, there is a great deal of variation in yield among different templates. If the yield is low, the first step in troubleshooting the reaction is to use the Control Template in a standard MEGAscript RNAi Kit transcription reaction to determine if the problem is with the template, the reagents,or procedure.
Neither my template nor the control reaction works
If the positive control does not work, it could be an indication that something is wrong with the kit; call Ambion’s Technical Support group for more troubleshooting help.
The control reaction works,but my template gives low yield
Figure 5
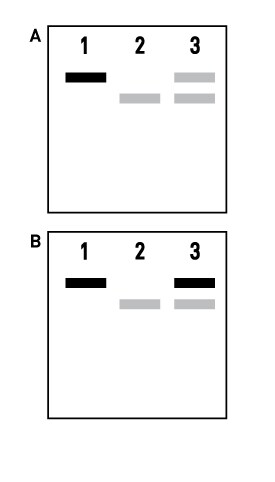
If the transcription reaction with your template generates full-length, intact RNA, but the reaction yield is significantly lower than the amount of RNA obtained with the Control Template, it is possible that
contaminants in the DNA are inhibiting the RNA polymerase. A mixing experiment can help to differentiate between problems caused by inhibitors of transcription and problems caused by the sequence of a template. Include three reactions in the mixing experiment using the following DNA templates:
1. 2 μL Control Template
2. 1–2 μg experimental DNA template
3. a mixture of 1 and 2
Assess the results of the mixing experiment by running 0.5 μL of the transcription reaction on an agarose gel.
Transcription of the Control Template is inhibited by your template. (See Figure 6.a)
This implies that inhibitors are present in your DNA template. Typical inhibitors include residual SDS, salts, EDTA, and RNases. Proteinase K treatment followed by phenol:chloroform extraction frequently improves template quality. Proteinase K treatment Treat template DNA with Proteinase K (100–200 μg/mL) and SDS
(0.5%) for 30 min at 50°C, followed by phenol/chloroform extraction and ethanol precipitation. Carry-over of SDS can be minimized by diluting the nucleic acid several fold before ethanol precipitation, and excess salts and EDTA can be removed by vigorously rinsing nucleic acid pellets with 70% ethanol before resuspension.
Adding your template to the reaction with the Control Template does not inhibit synthesis of the control RNA.
(See Figure 6.b.) This indicates that the problem may be inherent to your template.
Check the amount and quality of template Template quantitation may be inaccurate. If quantitation was based on UV absorbance and the DNA prep had substantial amounts of RNA or chromosomal DNA, the amount of template DNA may be substantially less than the calculated value.
Also, check an aliquot of the template DNA on an agarose gel to make sure it is intact and that it is the expected size. If there is even a small amount of circular template in the transcription reaction it will reduce the yield of dsRNA (see section Plasmid linearization).
Extend the reaction time. Another parameter that can be adjusted is reaction time. Extending the standard 2–4 hr incubation to 6–10 hr,
Change your priming region.
Some sequences are simply inefficient transcription templates. If you get low RNA yields, even after checking the template and trying overnight incubation of the transcription reaction, it may be necessary to prepare a transcription template from a different region of the gene. Often simply moving the transcription start point can overcome problems with inefficient transcription; typically there are several regions of the gene that will transcribe with equal efficiency. Also, transcription efficiency may be higher when the transcription template contains 2–3 bases of purines immediately following the GGG sequence at positions +1 to +3 in the T7 promoter sequence • Use 2 separate templates Sometimes the template will simply not transcribe well with opposing promoters. In this case, the two strands of RNA need to be made from separate templates and annealed after synthesis
C. Multiple Reaction Products, Transcripts of the Wrong Size
Reaction products produce a smear when run on a gel
This problem is usually seen with single-strand transcriptions. If the RNA appears degraded (e.g. smeared), remove residual RNase from the DNA template preparation before in vitro transcription. Do this by digesting the DNA prep with proteinase K. The RNase Inhibitor in the transcription reaction can only inactivate moderate RNase contamination. Large amounts of RNase in the DNA template will compromise the size and amount of
transcription products.
Reaction products run as more than one band, or as a single band smaller than expected
Premature termination of transcription
If gel analysis shows multiple discrete bands or a single band smaller than the expected size, there may be problems with premature termination by the polymerase. Even if transcription of only one of the strands was prematurely terminated, the single-stranded portion of the duplex will be digested during the nuclease treatment, resulting in a shorter than expected dsRNA.
Possible causes of premature termination are sequences which resemble the phage polymerase termination signals, stretches of a single nucleotides, and GC-rich templates.
Termination at single polynucleotide stretches can sometimes be alleviated by decreasing the transcription reaction temperature (Krieg 1990). We suggest testing incubations at 30°C, 20°C and 10°C, but be sure to increase the reaction time to offset the decrease in yield caused by incubation at suboptimal temperatures.
There is a report that single-stranded binding (SSB) protein increased the transcription efficiency of a GC rich template (Aziz and Soreq, 1990).
Reaction products are larger than expected
Products occasionally run as two bands after the nuclease digestion and dsRNA purification; one at the expected size, and one that is double the expected size. If this occurs, check the size of the transcription template on a gel to verify that it is pure and sized correctly. dsRNA that contains a double-sized band can be used for RNAi with no problems, in fact double-sized bands are sometimes seen from the Control Template
Multi-strand aggregates are present in the mixture
Larger than expected bands or ethidium bromide staining in the wells could be seen as a result of aggregates of multiple RNA strands. These can be denatured by heating the solution to 75–100°C for ~3 min, then allowing it to cool to room temperature. Be sure that RNA is in a solution containing at least 1 mM EDTA (such as the Elution Solution supplied with the kit) for the heat treatment.
- Aziz RB and Soreq H (1990) Improving poor in vitro transcription from GC-rich genes. Nucl. Acids Res.
18:3418. - Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, and Mello CC. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391(6669):806–11.
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, and Ahringer J (2001) Effectiveness of specific
RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome
Biol.2(1):RESEARCH 0002. - Krieg PA (1990) Improved Synthesis of Full-Length RNA Probes at Reduced Incubation Temperatures. Nucl. Acids Res. 18: 6463.
- Lohmann JU, Endl I and Bosch TC (1999) Silencing of developmental genes in Hydra. Dev Biol.
214(1):211–4. - Milligan JF, Groebe DR, Witherell GW, and Uhlenbeck OC (1987) Oligoribonucleotide synthesis using T7
RNA polymerase and synthetic DNA template. Nucl. Acids Res. 15: 8783–8798. - Misquitta L and Paterson BM (1999) Targeted disruption of gene function in Drosophila by RNA interference
(RNA-i): a role for nautilus in embryonic somatic muscle formation. Proc Natl Acad Sci USA. 96(4):1451–6. - Morris JC, Wang Z, Drew ME, Paul KS, and Englund PT (2001) Inhibition of bloodstream form Trypanosoma brucei gene expression by RNA interference using the pZJM dual T7 vector. Mol Biochem Parasitol. 117(1):111–3.
- Ngo H, Tschudi C, Gull K, and Ullu E (1998) Double-stranded RNA induces mRNA degradation in Trypanosoma
brucei. Proc Natl Acad Sci USA 95(25):14687–92. - Rubin GM and Spradling AC (1982) Genetic transformation of Drosophila with transposable element vectors. Science 218(4570):348–53.
- Sanchez Alvarado A and Newmark PA (1999) Double-stranded RNA specifically disrupts gene expression during planarian regeneration. Proc Natl Acad Sci USA 96(9):5049–54.
- Schenborn ET and Mierendorf RC (1985) A novel transcription property of SP6 and T7 RNA polymerases:
dependence on template structure. Nucl. Acids Res. 13: 6223–6236. - Timmons L and Fire A (1998) Specific interference by ingested dsRNA. Nature 395(6705):854.
- Wargelius A, Ellingsen S, and Fjose A (1999) Double-stranded RNA induces specific developmental efects in zebrafish embryos. Biochem Biophys Res Commun. 263(1):156–61.
- Zamore PD, Tuschl T, Sharp PA, and Bartel DP (2000) RNAi: double-stranded RNA directs the ATP-dependent
cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101(1):25–33.
1626M Revision B: Oct 2009