In the last seven years that Chromatin Immunoprecipitation (ChIP) sequencing (ChIP-Seq) has been around, it has become the technique for showing us where transcription factor binding regions are found on chromosomes genome-wide. What’s important about that? Oh, just that these regions where proteins and DNA interact are essential to a number of biological processes like cell division and differentiation, gene regulation, nucleosome positioning, and epigenetic functions like histone and chromatin modification.
The older method, chromatin immunoprecipitation by microarray chip analysis, required more input material, was subject to higher variability, and was technically much more difficult to perform.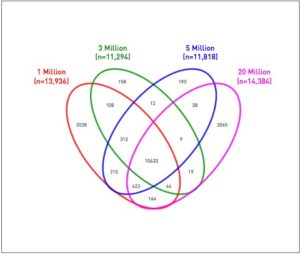
ChIP-Seq has not only shown us how genetic and epigenetic information is regulated and transferred onward, it also can show us what happens when something – usually disease – goes wrong with these processes which is usually associated with disease..
But ChIP-Seq’s usefulness has been hampered in the past because often the technique requires a lot of cellular DNA. The older method’s hunger for cells meant that only highly abundant cell types were realistically available for ChIP sequencing. Small populations of cell typesjust weren’t a part of this program.
ChIP-Seq works with small populations
Scientists at Thermo Fisher Scientific, however, optimized a ChIP-Seq protocol which doesn’t require as many cells, or as much DNA to allow these smaller cell populations to fall within the grasp of ChIP-Seq analysis. In addition, results were comparable to results obtained using higher cell numbers by ENCODE (the Encyclopedia of DNA Elements), a project to systematically map transcription regions, transcription factor binding, chromatin structure and histone modifications.
ChIP-Seq methods enrich and map binding sites for transcription factors, epigenetic contacts and other protein-DNA touchpoints. First, cells are treated with a fixative like formaldehyde, the DNA and associated proteins making up chromatin are cross linked, and antibody binding (immunoprecipitation) to enrich for bound DNA is done. Then, that DNA is sequenced, mapped to the genome, and significant peaks are called from that aligned reads.
The optimized protocol was specifically designed for the Ion Proton™ System. Our scientists focused on a control cell line, K-562, which we analyzed for genomic binding sites occupied by the transcription factor CTCF, histone activation modification H3K27ac, and histone silencing modification H3K27me3. After enrichment with the MAGnify™ Chromatin Immunoprecipitation System, the libraries were created and barcoded for sequencing on the Ion Proton™ System.
Multiplexing four samples on an Ion PI™ v2 Chip resulted in 89.4 and 89.7 million reads with an excellent barcode balance of 19.4–25.8 million reads per sample aligned to the human genome. Highly concordant results with ENCODE data were seen using only 1 million cells.
A closer look: Comparing dendritic cell results
Dendritic cells (DCs) are antigen-presenting cells help regulate immune system functions. To provide a diversity of regulatory functions, DCs differentiate from bone marrow stem cells into multiple different types or lineages. These different lineage pathways are controlled through transcription factors like Irf4, which is needed for proper CD11b+ DCs lineage development. Malfunctions in this development or the function of CD11b+ DCs have been linked to various inflammatory diseases including asthma. Due to limited cell numbers, the epigenetic connections between CD11b+ immune cell specialization and inflammatory disease are virtually unknown.
To address the challenges of limited sample input and to test the utility and robustness of the optimized ChIP-Seq protocol, ChIP with antibodies to Irf4 was used to determine the genomic binding sites present using flow cytometry sorted populations of 1, 3, 5, or 20 million mouse DCs of the CD11b+ lineage. Using Ion Torrent sequencing of ChIP DNA, a team from Thermo Fisher Scientific and National Jewish Health Integrated Center for Genes, Environment & Health in Denver created an unbiased, genome-wide view of transcription factor binding sites of Irf4. This analysis identified Irf4 gene targets that were expressed in CD11b+ as well as DC103+ dendritic cells, which may constitute a regulation program for Irf4’s control of dendritic cell lineage development. The researchers found high agreement, with 61% (n = 10,632) out of a total of 17,417 significant peaks, that were in common across all cell inputs. Successful ChIP-Seq analysis of low cell number inputs using the Ion Proton™ System indicate that the discovery of regions and associated with Irf4 binding are concordant with larger cell number inputs.
A ChIP-Seq in every lab
Precious samples include primary cells, stem cells, and biopsied material, and the ability to perform ChIP-Seq on them opens the door to understanding how cell begin differentiation and develop key regulatory processes, as well as how specific cell populations may contribute to disease. This nearly ubiquitous technique now has the potential to reach further into the lab, and into our understanding of cell biology.
More information about Ion Torrent Chromatin ImmunoPrecipitation Sequencing (ChIP-Seq) can be found here.
Leave a Reply