Search
Characterization of miRNA Expression in Normal Human Tissues
Measuring the amount of naturally-occurring miRNAs in tissues of different physiological and pathological conditions is an important first step to investigate the functions of miRNAs. Applied Biosystems scientists examined the profiles of 345 human miRNAs in 40 normal tissues using TaqMan MicroRNA Assays. These profiles provide a baseline against which miRNA profiles in tissues can be compared.
Normalizing RNA Input
The expression of 345 human miRNAs was quantitated in 40 normal human tissues including brain, muscle, circulatory, respiratory, lymphoid, gastrointestinal, urinary, reproductive, and endocrine systems. Four human miRNAs (miR-30e, miR-92, miR-92N, and miR-423) were identified as the least variable across this study. The average value of these miRNAs was used to normalize the RNA input between tissues. Sample input was also normalized by quantitating small nuclear RNAs using the TaqMan MicroRNA Assay Controls, and the key patterns of the hierarchical clustering of both miRNAs and tissues was very similar.
Hierarchical Clustering
Normalized data from assays were subjected to hierarchical clustering using two methods: 1) mean-centering data for each miRNA but not tissues, followed by correlation similarity metrics for both miRNA and tissue clustering; and 2) Euclidean similarity metric and correlation similarity metric to cluster miRNAs and samples, respectively, without centering the data.
miRNA Profiles Correlate with Anatomically and Physiologically Associated Tissues
Unsupervised hierarchical clustering based on expression variation for each miRNA across the specimens was used to examine the correlation between different tissue types. In general, normal human tissues derived from similar anatomical locations or, with related physiological functions, were primarily clustered together. For example, samples from different locations in the heart (atrium versus ventricle) were clustered with skeletal muscle. This was also true for gastrointestinal tissues (stomach, small intestine, and colon), lymphoid tissues (spleen and lymph node), female reproductive organs (ovary, uterus, and cervix), and respiratory tissues (lung and trachea) (Figure 1). This result is consistent with the published clustering patterns of normal tissues using mRNA expression profiles [1].
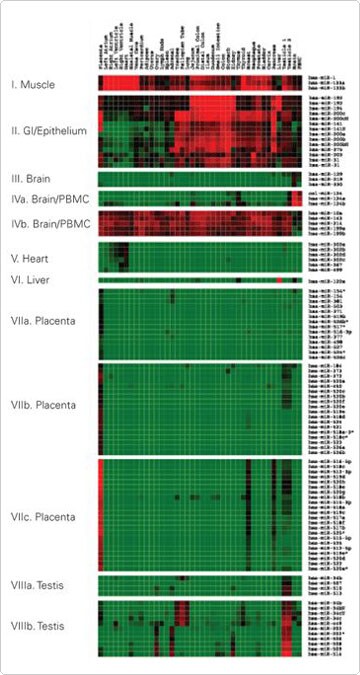
Figure 1. Unsupervised Hierarchical Clustering of Normal Human Tissues Based on the Variation of miRNA Abundance. The pseudocolor scale outlines the CT values represented in the heat map. Reproduced with permission from BMC Genomics 2007, 8:166 [2].
Figure 1. Unsupervised Hierarchical Clustering of Normal Human Tissues Based on the Variation of miRNA Abundance. The pseudocolor scale outlines the CT values represented in the heat map. Reproduced with permission from BMC Genomics 2007, 8:166 [2].
Differentially Expressed Groups Clustered in Genome
Eight miRNA groups were identified based on the correlation coefficient (r>0.9) between the miRNA expression patterns within each group. The miRNAs in some of the eight, differentially expressed groups appeared to be localized within the same genomic region within 1–5 kb from each other. Almost all miRNAs located in the same genomic clusters were overrepresented in the differentially expressed groups as determined by Chi-square tests. Two examples are miRNAs in groups VII and VIII. These miRNAs were preferentially expressed in placenta and testes and are localized in two separate genomic clusters at chromosomes 19q13.42 and Xq27.3, respectively (Figure 2).
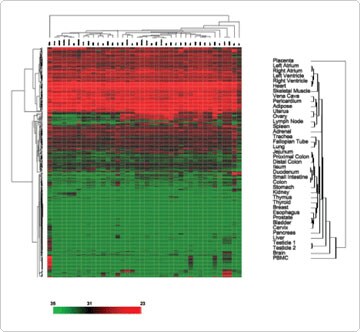
Figure 2. An Enlarged View of the Eight Groups of Most Differentially Expressed miRNAs. Reproduced with permission from BMC Genomics 2007, 8:166 [2].
Figure 2. An Enlarged View of the Eight Groups of Most Differentially Expressed miRNAs. Reproduced with permission from BMC Genomics 2007, 8:166 [2].
Preferential Expression
Fifteen miRNAs were consistently expressed in all tissues, making them strong candidates for normalizing controls. Most differentially expressed miRNAs were divided into two categories: those with preferential expression in tissues having similar functions or structures, and those with specific expression, or lack of expression, in certain tissues.
miR-129, miR-219, and miR-330, showed specific expression in brain. The computationally predicted targets for these miRNAs showed reduced expression in brain, while their expression in non-brain tissues was highly variable. miR-199a, miR-199b, and miR-214 showed low expression in brain and peripheral blood mononuclear cells (PBMC) relative to the other tissues. Computational predictions of brain and PBMC miRNA targets indicate expression in one or the other tissue, but not both.
These results indicate that miRNA expression may be controlled by a binary effect on expression of its targets: when miRNA expression is high, target gene expression is low; and when miRNA expression is low, expression of the target genes may be controlled by other elements such as transcription factors.
Scientific Contributors
Yu Liang, Dana Ridzon, Linda Wong, Caifu Chen • Applied Biosystems, Foster City, CA
miR-129, miR-219, and miR-330, showed specific expression in brain. The computationally predicted targets for these miRNAs showed reduced expression in brain, while their expression in non-brain tissues was highly variable. miR-199a, miR-199b, and miR-214 showed low expression in brain and peripheral blood mononuclear cells (PBMC) relative to the other tissues. Computational predictions of brain and PBMC miRNA targets indicate expression in one or the other tissue, but not both.
These results indicate that miRNA expression may be controlled by a binary effect on expression of its targets: when miRNA expression is high, target gene expression is low; and when miRNA expression is low, expression of the target genes may be controlled by other elements such as transcription factors.
Scientific Contributors
Yu Liang, Dana Ridzon, Linda Wong, Caifu Chen • Applied Biosystems, Foster City, CA
TaqMan MicroRNA AssaysTaqMan MicroRNA Assays quantitate miRNAs with the specificity and sensitivity of TaqMan assay chemistry. A simple, two-step protocol requires only reverse transcription with a miRNA-specific primer, followed by real-time PCR with TaqMan probes. The assays target only mature miRNAs, not their precursors, ensuring biologically relevant results. TaqMan MicroRNA Assays are available for human, mouse, rat, Arabidopsis, C. elegans, and Drosophila samples.
Each kit contains oligonucleotides for 50 RT reactions and 150 TaqMan reactions.
Each kit contains oligonucleotides for 50 RT reactions and 150 TaqMan reactions.
References1. Shyamsundar R, Kim YH, Higgins JP, Montgomery K, Jorden M, Sethuraman A, van de Rijn M, Botstein D, Brown PO, Pollack JR (2005) A DNA microarray survey of gene expression in normal human tissues. Genome Biology 2005, 6:R22
2. Liang Y, Ridzon D, Wong L, Chen C (2007) Characterization of microRNA expression profiles in normal human tissues BMC Genomics 2007, 8:166
2. Liang Y, Ridzon D, Wong L, Chen C (2007) Characterization of microRNA expression profiles in normal human tissues BMC Genomics 2007, 8:166