在细胞治疗制造过程中,缺乏明确的监管框架常常导致原材料的选择和资格认定变得复杂且不清晰。在本博客文章中,我们将探讨在细胞治疗制造中选择原材料的关键考量,以及早期选择对于确保生产效率和最终治疗产品质量的重要性。
目录
- 为何早期材料策略决定临床成功
- 了解细胞治疗中的原材料与辅助材料
- 监管环境与GMP要求
- 定义GMP级别及供应商资质认定
- 评估关键原材料属性
- 文件记录与可追溯性要点
- 监管支持文件与协作
- 案例研究
- 摘要:制定长期原材料战略
完整故事请见,
为何早期材料策略决定临床成功
细胞治疗产品的临床转化成功依赖于早期对工艺设计和支持该工艺的原材料的决策。在工艺开发过程中选择的材料会直接影响生产效率、可扩展性、安全性以及最终的监管批准。如果原材料未能达到临床或商业级标准,在后续开发阶段进行替换可能导致昂贵的可比性测试、重新验证和监管延迟。
一种强调使用的有意识战略GMP级原材料或在以下条件下制造的材料:健全的质量管理体系从一开始就采取措施有助于降低这些风险。这种主动的方法支持合规监管,提高一致性,并增加从实验室到临床成功转化的概率。在下面的后续章节中,我们将进一步了解一些实际案例,这些案例展示了早期决策如何帮助确保风险缓解。
了解细胞治疗中的原材料与辅助材料
原材料,也称为辅助或辅助性材料,是在制造过程中与治疗产品接触但不打算保留在最终药品中的物质。这些包括细胞培养基,细胞因子、酶, enzymes, 缓冲液、赋形剂和冷冻保护剂。虽然它们会被从最终配方中去除,但其规格可能深刻影响最终细胞治疗产品的纯度、效力和安全性。下表列出了原材料风险评估的关键考虑因素。

全球监管机构,如FDA、EMA和WHO,已经为这些材料采用了互补的定义,并制定了相关框架,例如USP <1043>,欧洲药典 5.2.12,以及ISO 20399强调基于风险的合格评定。
全球最受认可的原材料指导文件之一USP <1043>,为根据相关风险将原材料分为四个不同等级提供了框架(见图1)。
这些指南共同要求治疗性产品开发者尽可能使用最高等级的材料——优选一级或二级风险类别根据USP <1043> — 并避免仅供研究使用(RUO)材料用于长期流程。
如需了解更多关于我们Gibco™细胞治疗系统™(CTS™)培养基和试剂的信息
监管环境与GMP要求
细胞治疗是医学中新兴的疗法,面对不断变化的监管环境可能令人望而生畏。对原材料进行基于风险的分层有助于指导和影响材料选择。在下文中,我们指出了一些重要的监管框架。

USP <1043> 和 ISO 20399 指南
该USP <1043> 细胞、基因和组织工程产品的辅助材料章节以及ISO 20399共同定义了基于风险的材料采购期望。这些框架强调通过评估供应商的制造过程控制能力,可追溯性,以及检测实践,而不是仅仅依赖于诸如“GMP级别”等市场宣传标签。
鼓励开发人员进行供应商风险评估,需考虑以下因素:
- 供应商质量认证(ISO 9001、ISO 13485、EXCiPACT GMP)
- 病毒安全性和内毒素检测数据
- 批次间差异及性能数据
- 来源和可追溯性的文件记录
需要注意的是,目前尚无全球统一定义的“GMP级”细胞治疗原材料。
定义GMP级别及供应商资质认定
由于没有通用的“GMP级”定义,开发人员必须通过审核、质量协议以及变更通知程序来验证供应商合规性。许多制造商现在采用ICH Q11和Q12生命周期控制框架,实现从研发、临床到商业阶段的一致性。
维护监管支持文件(RSFs)或药品主文件(DMFs)的供应商为下游开发者(细胞和基因治疗制造商)提供了高效的监管审查路径,减少在申报中披露专有信息的需求。
评估关键原材料属性
在制造的早期阶段,评估原材料的关键质量属性至关重要,例如性能、安全性、身份、纯度、一致性和稳定性。必要时应采用功能性细胞检测,以提供有关细胞活力或扩增能力等详细信息。
性能、安全性和一致性
- 每种原材料都应针对其性能(满足预期细胞培养功能的能力)、安全性(无外来因子)和一致性(批次间差异最小)进行评估。
- 参考测试方法,如美国药典(USP),对于合规、准确且相关的原材料评估与报告非常重要。USP 和 EP 级原材料具有明确药典标准,为身份、纯度和效力提供了标准化测试。
- 供应商还应展示定量且与应用相关的数据,例如用于 T 细胞培养基的 T 细胞扩增实验,以确保其相关性。
- 如果信息缺失或不充分,开发者可能需要使用经过验证或药典方法进行内部验证,以确保选择的可靠性。
生物风险与动物来源成分
降低生物风险仍然是首要任务。鼓励开发者使用无动物来源(AOF)或无异种成分尽可能使用这些材料。如果无法避免动物来源的成分,风险评估应考虑:
- 重组或合成替代品
- 病毒灭活(如伽马射线照射、过滤)
- 上游与下游使用(后期工艺阶段风险增加)
- 供应商可追溯性和原产地证明
- 原产国疯牛病/传染性海绵状脑病(BSE/TSE)相关考量
此类基于风险的策略与当前做法相一致FDA CMC 指南以及国际协调努力ICH Q9(质量风险管理).
请访问我们的网站,了解更多无动物源重组蛋白的信息
文件记录与可追溯性要点
全面的文件记录对于证明控制和合规至关重要。主要供应商文件包括:
- 分析证书(COA):详细说明测试结果、规格和验证方法
- 原产地证书(COO):确认生物可追溯性及无受限来源
- 安全与法规声明:概述GMP生产、无菌验证和检测
这些文件使开发者能够评估可比性并准备数据以供纳入通用技术文件(CTD)第3.2.S.2.3节(材料控制)。
Gibco™ CTS™免疫细胞血清替代品专为细胞治疗制造而设计,并满足可追溯性文件要求。它符合美国、欧洲和日本的原材料指导原则。该试剂旨在替代人血清用于执行体外人淋巴细胞培养。

想了解更多关于赛默飞世尔科技产品的信息,这些产品能够帮助提升您的流程效率,同时兼顾法规合规吗?

法规支持文件与协作合规
原材料供应商是法规合规的重要合作伙伴。他们的变更控制系统和沟通方式会显著影响开发者保持申报资料最新的能力。
最佳实践包括:
- 质量协议:明确变更通知时间表及所需文件。
- 供应商资格认证与审核:确保遵循ISO 20399和USP <1043>原则。
- 变更管理协调:将供应商发起的变更映射到监管分类(重大、中等、轻微)。
- QbD集成:定义关键材料属性(CMA)以及目标材料特性(TMP),以支持灵活且合规的生命周期管理。
这些方法以设计质量(QbD)为基础,采用科学依据的规范和基于科学的规范—帮助简化全球申报流程并降低供应风险。以下部分总结了两个案例研究,详细描述了这些最佳实践。
案例研究
案例研究1
例如,一家总部位于美国的药物开发商联系其供应商,为一种新疗法的全球产品注册使用化学定义的专有培养基。鉴于全球范围内(原材料)主文件在各地区的有限可用性,供应商的法规团队提供了一个申报后计划其中包括:(i) 信息请求和 (ii) 原材料变更管理。
药物开发商加入了供应商的变更通知项目,并与制造工厂正式签订了商业供应协议。这些措施确保所有申报后文件都得到充分合规和管理。
案例研究 2
在另一个例子中,一家为多种FDA批准的细胞治疗产品提供培养基的供应商正在扩建其生产基地。尽管两个地区的质量管理体系保持一致,但由于生产地点不同,关于培养基可比性的问题仍然存在。在这种情况下,供应商使用了II型主文件(Type II Master File)向FDA传达可比性数据,以确保产品供应不中断。
通常,II型主文件包含有关药品所用材料(包括细胞和基因治疗产品)的制造、表征、质量控制和稳定性的详细信息。
这些主文件用于关键原材料,例如细胞因子生长因子、培养基成分、酶和缓冲液等。这些材料可能不是最终药品的一部分,但会显著影响产品的质量、安全性和一致性。
摘要:制定长期原材料战略
成功的原材料战略不仅仅是初始采购决策——它还包括法规协调、供应商合作以及变更管理规划。通过采用全球统一的框架(USP <1043>、ISO 20399、ICH Q11/Q12),制造商可以为合规、灵活且可扩展的生产建立结构化基础。
这种前瞻性方法的关键要素列在下表:
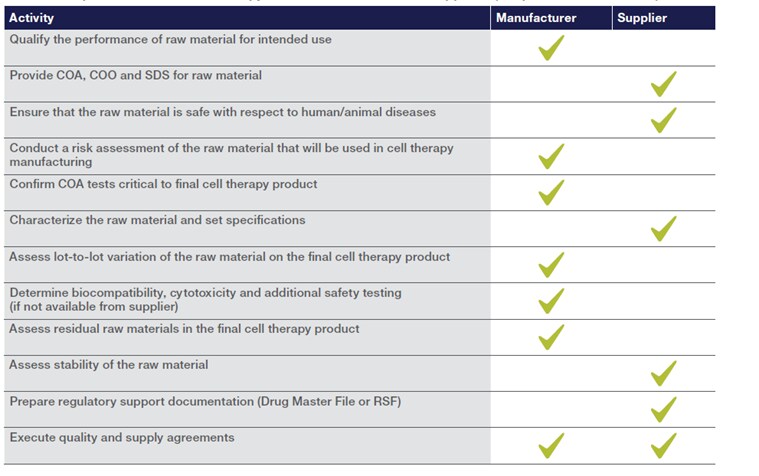
最终,这些实践促进了供应链韧性,并支持全球一致且合规的细胞治疗制造。
深入探讨细胞治疗开发中无污染、稳定且符合法规要求原材料的选择标准。
更多资源
- 宣传册:Gibco™ PeproGMP™ 细胞因子——面向未来的治疗制造
- 网络研讨会:在细胞和基因治疗监管申报中管理原材料变更
- 细胞治疗系统(CTS)
- Gibco PeproGMP 细胞因子与生长因子
- 赛默飞世尔科技:细胞治疗制造解决方案
主要要点总结
- 早期采用GMP级原材料提升合规性和产品可靠性。
- 应用USP <1043>,ISO 20399,以及ICH Q11/Q12用于基于风险的控制。
- 整合质量源于设计(QbD)原则和关键材料属性(CMA)转化为材料规范。
- 建立强有力的供应商协作和变更通知系统以确保生命周期合规。
- 将内部质量体系与不断变化的监管指导保持一致,以维护具有韧性、面向未来的供应链。
正在优化您的细胞治疗开发流程吗?探索用于细胞治疗制造的 PeproGMP 细胞因子。
用于细胞、基因或组织类产品的研究或生产。注意:不适用于直接给人或动物进行给药。
尾注或参考文献
- Kime K., Peng X. Ensuring compliance through collaboration: managing raw material changes in cell and gene therapy regulatory filings. Cell & Gene Therapy Insights. 2025; 11(4): 275–288.
- USP <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products.
- ICH Q11 and Q12 Guidelines.
- FDA Guidance for Industry: Chemistry, Manufacturing, and Control Information for Human Gene Therapy INDs.

siRNA与miRNA:解码这些微小却强大的分子
RNA干扰(RNAi)是一种高度保守且至关重要的�... Dana D'Amico
了解更多
显微图像去卷积:清晰显微镜指南
显微镜极大地提升了我们观察不可见世界的能... Dana D'Amico
了解更多
Dynabeads磁珠:技巧与窍门
在这个三篇博客系列的最后一篇中,Ketil Peders... Ketil Winther Pedersen
了解更多
多重免疫分析指南:多重ELISA及更多
多重免疫分析,包括多重ELISA,是研究人员同�... Dana D'Amico
了解更多
发表回复