In cell therapy manufacturing, the lack of definitive regulatory frameworks often makes the process of selecting and qualifying raw materials complex and unclear. In this blog post, we will explore the key considerations for choosing raw materials in cell therapy manufacturing and the importance of early selection in ensuring production efficiency and the quality of final therapeutic products.
Table of contents
- Why early material strategy defines clinical success
- Understanding raw and ancillary materials in cell therapy
- Regulatory landscape and GMP expectations
- Defining GMP-grade and supplier qualification
- Assessing critical raw material attributes
- Documentation and traceability essentials
- Regulatory Support Files and collaboration
- Case Studies
- Summary: Building a long-term raw material strategy
For the full story,
Why early material strategy defines clinical success
Successful clinical translation of a cell therapy product depends on early decisions about both the process design and the raw materials that support it. The materials selected during process development directly influence manufacturing efficiency, scalability, safety, and eventual regulatory approval. If raw materials fail to meet clinical- or commercial-grade standards, substitutions later in development can lead to costly comparability testing, revalidation, and regulatory delays.
An intentional strategy that emphasizes the use of GMP-grade raw materials or materials manufactured under robust quality management systems from the outset helps reduce these risks. This proactive approach supports regulatory compliance, improves consistency, and increases the probability of successful translation from bench to bedside. In a later section below, we will learn more about real-world examples of intentional strategies where early decisions helped ensure risk mitigation.
Understanding raw and ancillary materials in cell therapy
Raw materials, also referred to as ancillary or auxiliary materials,are substances that contact the therapeutic product during manufacturing but are not intended to remain in the final drug product. These include cell culture media, cytokines, enzymes, buffers, excipients, and cryoprotectants. Although they are removed from the final formulation, their specifications can profoundly affect the purity, potency, and safety of the final cell therapy. Table below lists the key considerations for raw material risk assessment.

Global regulatory bodies such as the FDA, EMA, and WHO have adopted complementary definitions for these materials, with frameworks like USP <1043>, Ph. Eur. 5.2.12, and ISO 20399 emphasizing risk-based qualification.
One of the most globally recognized raw material guidance documents, USP <1043>, provides a framework for classifying raw materials into four different tiers based on associated risks (Figure 1).
These guidelines converge on the expectation that therapeutic developers use the highest-grade materials feasible—preferably Tier 1 or Tier 2 risk categories per USP <1043> — and avoid Research Use Only (RUO) materials for long-term processes.
To learn more about our Gibco™ Cell Therapy Systems™ (CTS™) media and reagents
Regulatory landscape and GMP expectations
Cell therapies are newly emerging modalities in medicine, and navigating the ever-evolving regulatory landscape can be daunting. Risk-based stratification of raw materials helps guide and influence material selection. In the paragraph below, we note some important regulatory frameworks.

USP <1043> and ISO 20399 guidance
The USP <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products chapter and ISO 20399 together define risk-based expectations for material sourcing. These frameworks emphasize evaluating suppliers by their control of manufacturing processes, traceability, and testing practices, rather than relying solely on marketing designations such as “GMP-grade.”
Developers are encouraged to conduct supplier risk assessments that consider:
- Supplier quality certifications (ISO 9001, ISO 13485, EXCiPACT GMP)
- Viral safety and endotoxin testing data
- Lot-to-lot variability and performance data
- Documentation of origin and traceability
It should be noted that currently there are no globally defined “GMP-grade” raw materials for cell therapy manufacturing.
Defining GMP-grade and supplier qualification
Because no universal “GMP-grade” definition exists, developers must verify supplier compliance through audits, quality agreements, and change-notification programs. Many manufacturers now adopt ICH Q11 and Q12 frameworks for lifecycle control, enabling consistency across investigational, clinical, and commercial stages.
Suppliers who maintain Regulatory Support Files (RSFs) or Drug Master Files (DMFs) offer downstream developers (cell and gene therapy manufacturers) an efficient path for regulatory review, reducing the need to disclose proprietary information in filings.
Assessing critical raw material attributes
It is essential to evaluate raw materials for critical quality attributes, such as performance, safety, identity, purity, consistency, and stability in the early stages of manufacturing. Functional cell-based assays, when required, should be used to provide detailed information on cell viability, or expandability, for example.
Performance, safety, and consistency
- Each raw material should be evaluated for performance (ability to meet the intended cell-culture function), safety (absence of adventitious agents), and consistency (minimal lot-to-lot variability).
- Reference test methods such as United States Pharmacopeia (USP) are important for compliant, accurate and relevant assessment and reporting on raw materials. USP and EP-grade materials with defined monographs provide standardized tests for identity, purity, and potency.
- Suppliers should also demonstrate quantitative, application-relevant data, e.g., T-cell expansion assays for media intended for T-cell culture, to ensure relevance.
- If the information is lacking or inadequate, developers may need to conduct in-house verification using validated or compendial methods to ensure robust selection.
Biological risk and animal-origin components
Mitigating biological risk remains a top priority. Developers are encouraged to use animal-origin-free (AOF) or xeno-free materials wherever possible. When animal-derived components are unavoidable, risk assessments should consider:
- Recombinant or synthetic alternatives
- Viral inactivation (e.g., gamma irradiation, filtration)
- Upstream vs. downstream use (later process stages increase risk)
- Supplier traceability and Certificates of Origin
- Country-of-origin BSE/TSE considerations
Such risk-based strategies align with current FDA CMC guidance and international harmonization efforts under ICH Q9 (Quality Risk Management).
Visit our website for more information on animal-origin free recombinant proteins
Documentation and traceability essentials
Comprehensive documentation is central to demonstrating control and compliance. Key supplier documents include:
- Certificate of Analysis (COA): detailing test results, specifications, and validated methods
- Certificate of Origin (COO): confirming biological traceability and absence of restricted sources
- Safety and regulatory statements: outlining GMP manufacturing, sterility validation, and testing
These documents enable developers to evaluate comparability and prepare data for inclusion in Common Technical Document (CTD) sections 3.2.S.2.3 (Control of Materials).
Gibco™ CTS™ Immune Cell Serum Replacement is designed specifically for cell therapy manufacturing and meets traceability documentation requirements. It complies with the raw material guidance in the United States, Europe and Japan. The reagent is intended to replace the use of human serum when performing ex vivo culture of human lymphocytes.

Want to learn more about Thermo Fisher Scientific products that can help make your process more efficient, while keeping regulatory compliance in mind?

Regulatory Support Files and collaborative compliance
Raw material suppliers are key partners in regulatory compliance. Their change-control systems and communication practices can significantly affect a developer’s ability to maintain up-to-date filings.
Best practices include:
- Quality agreements: defining change-notification timelines and required documentation.
- Supplier qualification & audits: ensuring adherence to ISO 20399 and USP <1043> principles.
- Change management coordination: mapping supplier-initiated changes to regulatory classifications (major, moderate, minor).
- QbD Integration: defining Critical Material Attributes (CMAs) and target material profiles (TMPs) to support flexible yet compliant lifecycle management.
Such approaches, anchored in Quality by Design (QbD) and science-based specifications—help streamline global submissions and mitigate the supply risk. The following section summarizes two case studies that describe these best practices in detail.
Case study
Case Study 1
For example, a US-based drug developer contacted their supplier for global product registration of a new therapy using chemically defined proprietary media. Given the limited regional availability of (raw material) master files worldwide, the supplier’s regulatory team provided a post-submission plan that included: (i) request to information and (ii) change management of raw materials.
The drug developer enrolled in the supplier’s change notification program and formalized a commercial supply agreement with the manufacturing site. These measures ensured that all post-submission documents were fully compliant and managed.
Case Study 2
In another example, a cell culture media supplier providing media used in several FDA-approved cell therapy products, was expanding their manufacturing site. Although the quality management systems in both regions were aligned, there were questions around media comparability based on different sites. In this case, the supplier used a Type II Master File to communicate comparability data to the FDA, ensuring uninterrupted product availability.
Typically, a Type II Master File contains detailed information about the manufacture, characterization, quality control, and stability of materials used in drug products, including cell and gene therapies.
These are used for critical raw materials, such as cytokines, growth factors, media components, enzymes, and buffers, etc. These materials may not be part of the final drug product but can significantly impact product quality, safety, and consistency.
Summary: Building a long-term raw material strategy
A successful raw material strategy extends beyond initial sourcing decisions—it encompasses regulatory alignment, supplier partnerships, and change-management planning. By adopting globally harmonized frameworks (USP <1043>, ISO 20399, ICH Q11/Q12), manufacturers can create a structured foundation for compliant, flexible, and scalable production.
Key elements of this forward-looking approach are listed in the table below:
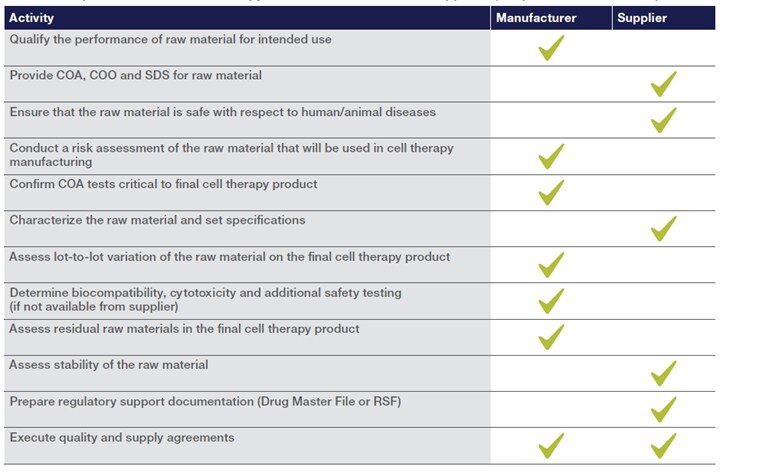
Ultimately, these practices foster resilient supply chains and support consistent, compliant cell therapy manufacturing globally.
Take a deep dive into selection criteria for contamination-free, stable, and regulatory-compliant raw materials in cell therapy development.
More resources
- Brochure: Gibco™ PeproGMP™ Cytokines: Future-Ready Therapeutics Manufacturing
- Webinar: Managing Raw Material Changes in Cell and Gene Therapy Regulatory Filings
- Cell Therapy Systems (CTS)
- Gibco PeproGMP Cytokines and Growth Factors
- Thermo Fisher Scientific: Cell Therapy Manufacturing Solutions
Key takeaways
- Early adoption of GMP-grade raw materials enhances compliance and product reliability.
- Apply USP <1043>, ISO 20399, and ICH Q11/Q12 for risk-based control.
- Integrate QbD principles and Critical Material Attributes (CMAs) into material specifications.
- Establish strong supplier collaboration and change-notification systems to ensure lifecycle compliance.
- Align internal quality systems with evolving regulatory guidance to maintain a resilient, future-ready supply chain.
Optimizing your cell therapy development process? Explore PeproGMP cytokines for Cell Therapy Manufacturing.
For Research Use or Manufacturing of Cell, Gene, or Tissue-Based Products. CAUTION: Not intended for direct administration into humans or animals.
Endnotes or References
- Kime K., Peng X. Ensuring compliance through collaboration: managing raw material changes in cell and gene therapy regulatory filings. Cell & Gene Therapy Insights. 2025; 11(4): 275–288.
- USP <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products.
- ICH Q11 and Q12 Guidelines.
- FDA Guidance for Industry: Chemistry, Manufacturing, and Control Information for Human Gene Therapy INDs.

Scaling Human-Relevant Oncology Testing: OncoPro Tumoroids on ScreenIn3D’s UpScale Platform
The United States National Institutes of Health has prioriti...
Read More
Why Most Modern Drug Candidates Fail at Solubility
Drug discovery capabilities continue to advance rapidly. Med... Dirk Leister
Read More
Scale-Up vs. Scale-Out Strategies for Continuous Pharma Manufacturing
As pharmaceutical pipelines evolve, manufacturing strategies... Dirk Leister
Read More
Cryopreservation: Advancements in Human Stem Cell-Derived Neurons
The field of cryopreservation has seen advancements, particu...
Read More
Leave a Reply