 Data acquisition and processing form the backbone of mass spectrometric (MS) proteomic analysis for accurate peptide identification and quantitation. The two strategies currently used in MS-based proteomics—data-dependent acquisition (DDA) and data-independent acquisition (DIA)—each present different benefits and drawbacks to the experimental design. In their recent paper, Prakash et al. (2014) present pSMART, a hybrid data acquisition and processing strategy that uses both strategies in tandem, giving enhanced results in terms of peptide identification and resulting protein quantitation.1
Data acquisition and processing form the backbone of mass spectrometric (MS) proteomic analysis for accurate peptide identification and quantitation. The two strategies currently used in MS-based proteomics—data-dependent acquisition (DDA) and data-independent acquisition (DIA)—each present different benefits and drawbacks to the experimental design. In their recent paper, Prakash et al. (2014) present pSMART, a hybrid data acquisition and processing strategy that uses both strategies in tandem, giving enhanced results in terms of peptide identification and resulting protein quantitation.1
The researchers used human plasma as the source material. Following standard trypsin digestion and subsequent spiking with peptide retention time calibration (PRTC) peptides (Thermo Scientific), they analyzed the undepleted sample using a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer coupled with an EASY-nLCII liquid chromatograph (both Thermo Scientific). Prakash and co-authors ran three replicates of each sample to establish parameters for experimental consistency.
The scientists operated the mass spectrometer in the three different acquisition modes. In DDA mode, they acquired tandem mass (MS/MS) spectra with higher-energy C-trap dissociation (HCD) fragmentation. In DIA mode, they ran the system in looped product ion mode. For pSMART analysis, they acquired both MS and DIA data independently, operating the system for high-resolution accurate-mass (HRAM) MS scanning.
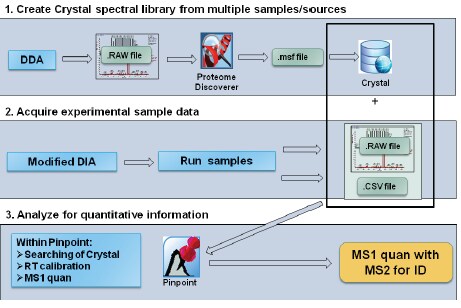
For spectral library searching, the research team used DDA experimental data of human plasma samples, acquired over two years from an LTQ Orbitrap Velos hybrid ion trap-Orbitrap mass spectrometer (Thermo Scientific) and the Q Exactive instrument. Using Crystal software (Thermo Scientific), they created the common spectral library from these data for subsequent evaluation of proteomic data generated from the three acquisition modes. From this strategy, and in comparison with an existing human protein database, the researchers could identify peptide fragments and evaluate abundance, comparing pSMART data acquisition and processing with the DDA and DIA mode workflows.
Following statistical processing and analysis of the results, Prakash et al. showed that their novel hybrid data acquisition method, pSMART, presented an enhanced workflow for proteomics identification and quantitation. Compared with results obtained from the DDA and DIA workflows, pSMART maintained experimental reproducibility in addition to identifying greater numbers of peptides. The authors are confident that pSMART combines the benefits of both acquisition modes to deliver greater sensitivity in a data processing tool.
Reference
1. Prakash, A., et al. (2014, September) “Hybrid data acquisition and processing strategies with increased throughput and selectivity: pSMART analysis for global qualitative and quantitative analysis,” Journal of Proteome Research, doi: 10.1021/pr5003017 [e-pub before print].
Post Author: Amanda Maxwell. Mixed media artist; blogger and social media communicator; clinical scientist and writer.
A digital space explorer, engaging readers by translating complex theories and subjects creatively into everyday language.

Where’s the Squeeze? Informatics Bottlenecks in Missing Protein Identification
The Chromosome-centric Human Proteome Project (C-HPP), an in...
Read More
Differential Protein Quantitation at the Peptide Level
Instead of quantifying differential expression changes in pr...
Read More
Post-Stroke Depression Likely Linked to Unresolved Inflammatory Response
The statistics associated with stroke are stark: of the 15 m...
Read More
Crunch Next-Generation Mass Spectrometric Proteomic Data without Running out of Memory
Increasing complexity of raw spectral data resulting from ta...
Read More
Leave a Reply