Search

Related Product Information
Go to...
Validation packets showing optimization of individual kinases in the LanthaScreen Kinase Activity Assay and LanthaScreen Eu Binding Assay are available. These validation packets show full assay optimization, including determining the ATP Km for LanthaScreen Activity Assays and tracer Kd determination for LanthaScreen Eu Kinase Binding Assays.
FAQ Contents
Filter settings, instruments, and plates
How do I set up LanthaScreen assays on my instrument?
Proper instrument setup and correct filter and mirror selection are crucial for success with LanthaScreen assays.
Conditions used for Eu-based LanthaScreen assays are not identical to other TR-FRET Eu-based assays.
Instrument-specific step-by-step setup guides for Tb- and Eu-based assays. If you do not see your plate reader, please contact us directly by email at drugdiscoverytech@lifetech.com.
Can I use a monochromator-based instrument to read LanthaScreen assays?
In general, filter-based instruments significantly outperform monochromator-based instruments for either Tb- or Eu-based LanthaScreen assays, unless the instrument has individually adjustable bandwidths for each emission, such as the Tecan Infinite M1000.
Are there specific plates I should use for my LanthaScreen assay?
Assays are typically performed in black, low-volume 384-well plates (Corning Part # 3676). White plates may yield higher-quality data (Corning Part # 3673) for assays that give a relatively low assay window and are essential for assays that are being measured on monochromator-based instruments or older filter-based instruments.
Understanding TR-FRET ratiometric data
The following three sections address common questions involving ratiometric data. These questions are:
- Why is it necessary to analyze the data as a ratio? Isn’t using the RFU easier?
- Why are the units of the ratio so low?
- Why does my assay window end at a value very low or less than 1.0?
- Is a bigger assay window better?
- Does an assay window of a certain size always mean my assay worked?
- Why should I calculate the emission ratio?
Why should I calculate the emission ratio?
Two emission signals are collected for TR-FRET assays, and taking a ratio of them represents a best practice in data analysis for TR-FRET assays including LanthaScreen assays and greatly increases assay reproducibility over reading the acceptor signal alone. The emission ratio is calculated by dividing the acceptor signal by the donor signal (520 nm/495 nm for Tb, and 665nm/615nm for Eu). Most Tb or Eu donors are not involved in producing TR-FRET signal and so the donor signal serves as a useful internal reference in the assay. Dividing by the donor signal will help account for small variances in the delivery (i.e., the pipetting) of the LanthaScreen reagents and lot-to-lot variability of the reagents.
TR-FRET is a distance-dependent phenomenon between donor and acceptor, and small differences in the number of labels or their positioning (donor or acceptor) from lot to lot can lead to differences in the acceptor output, 520 nm for Tb or 665 nm for Eu. The ratio is typically less affected by such lot-to-lot variation (Figure 16).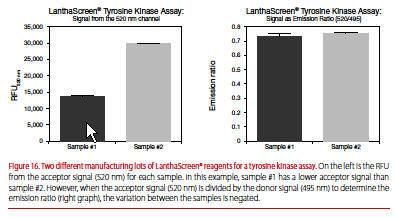
The numerical values of RFU are typically in the thousands or tens of thousands and these units fall away when the ratio is calculated. The statistical significance of the data is not affected. Some instruments arbitrarily multiply the ratio by 1,000 or 10,000 to make the units on the output look more familiar.
How do I calculate the response ratio?
Calculating a ratio of acceptor emission to donor emission RFU represents a best practice in data analysis for TR-FRET assays including LanthaScreen assays. Because the underlying donor and acceptor signals are dependent on instrument settings (such as instrument gain), the TR-FRET emission ratio and the resulting “top” and “bottom” of an assay window also will depend on these settings and vary from instrument to instrument. Numbers look quite small compared to relative fluorescence units (RFU) as a result of calculating the ratio.
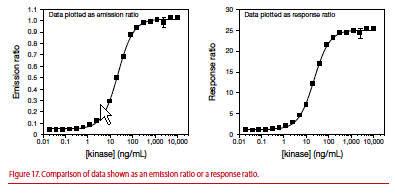
You can normalize the data in titration curves by dividing all values in the curve by the ratio obtained at the bottom of the curve to calculate a response ratio. In Figure 17, the value from each data point from the emission ratio graph (left) was divided by the emission ratio from the bottom of the curve (e.g., 0.05) and then plotted on the right as the response ratio beginning at 1.0. Use of a response ratio plot allows quick determination of the assay window and has no effect on IC50 values or Z’-factor.
Z’-factor: Is a 30-fold window better than a 10-fold window?
Assay window depends on instrument type and settings. What is important in determining the robustness of an assay is not the size of the window, but the size of error (standard deviation) in the data relative to the assay window. The “Z’-factor” value proposed by Zhang and colleagues [1] factors both of these into account and is a key metric to assess data quality in a TR-FRET assay. Assays with Z’ >0.5 are considered suitable for screening.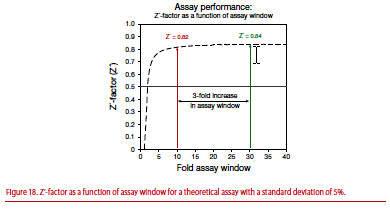
Figure 18 shows a theoretical graph of Z’-factor (Z’) as a function of assay window assuming a standard deviation of 5%. As the assay window increases, the Z’-factor quickly reaches a plateau within the first 4- to 5-fold increase in assay window. Above a 5-fold assay window, large changes in the fold assay window result in only an incremental increase in the Z’-factor. For example, with a 5% standard error, a 10-fold assay window results in a Z’-factor of 0.82. With a 3-fold increase in the assay window to 30, the observed gain in the Z’-factor is only 0.02. Statistically, the assay with the 10-fold window is just as suitable for screening as the 30-fold window.
1. Zhang JH, Chung TD, Oldenburg KR (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4(2):67–73.
Assay window alone is not a good measure of assay performance
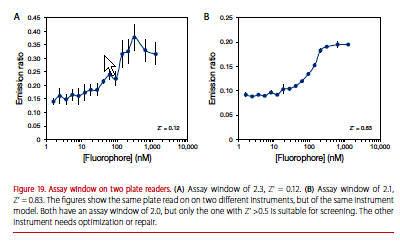
See the emission ratios for the same assay plate run on two different instruments (Figure 19). The instrument used to generate the curve on the right is suitable for the assay, but the one used to generate the curve on the left is not. Both have an assay window of 2.0, but the one on the left has more variability.
Assay conditions
How many assays can I run with a given amount of antibody?
This depends on the volume of the assay and the concentration of antibody used. At 2 nM antibody in a final volume of 20 μL, 25 μg of antibody will run >4,000 wells, and 1 mg of antibody will run >165,000 wells.
How many assays can I run with a given amount of substrate?
It varies, depending on the concentration of substrate used in the assay. Typically for the peptide substrates, 1 mg of peptide will run approximately 250,000 wells (10 μL reaction, 200 nM peptide). For Poly-GT or Poly-GAT substrates, the 1 mL, 30 μM size (~1 mg) will run approximately 16,700 wells (10 μL reaction, 200 μM substrate).
20 nmol of our physiological protein substrates is sufficient for approximately 10,000 wells (10 μL reaction, 400 nM substrate).
Chemical interferences to Tb or Eu chelate signals
Are the LanthaScreen reagents stable to interference from Mg2+, Mn2+, EDTA, and Fe3+?
The Tb or Eu chelate is completely stable to Mg2+. Mn2+ strongly competes with Tb3+ or Eu3+ for the chelate, resulting in loss of donor signal. However, interference by Mn2+ can be overcome by the presence of EDTA. The chelate is stable to levels of EDTA well beyond the concentration ranges used to stop kinase reactions (Figure 20).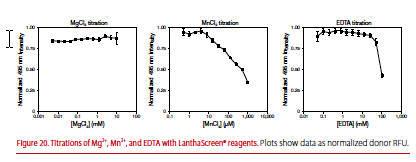
EDTA is added to stop kinase reactions and is typically mixed with the LanthaScreen antibody. An equimolar amount or slight excess of EDTA relative to the sum of Mn2+ and Mg2+ should be added with the antibody. In addition to using EDTA, using ratiometric data (division of the acceptor signal by the donor signal) compensates for any loss of signal caused by Mn2+(Figure 21).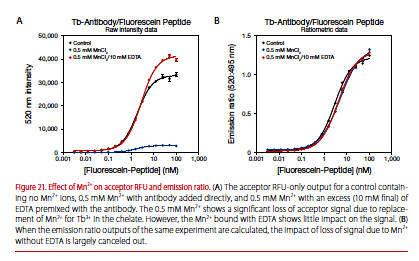
Fe3+ strongly competes with Tb 3+ or Eu3+, and will cause irreversible loss of donor signal.
Go to...
LT129 1-Jan-2010
For Research Use Only. Not for use in diagnostic procedures.