So you think the limit of denovo detection for Sanger is 20%? WRONG! We’ve been able to detect 5% minor variants at a sensitivity above 96% and specificity above 99.8% over an independent database (data not used in developing the methods) of 987 samples having 1112 true variants (constructed by dilution series), 895373 non-variant base positions, from 35 distinct amplicons taken from among the genes P53, BRAF, EGFR, HLA DPB1, HLA DQB1, KRAS, FLT3, RB1, CDG1, and ERB2.
BUT THERE’S MORE! With similar data we have been able to recover 2.5% and 1.25% variants.
The main trick is to minimize the noise underlying Sanger sequencing data using data from a control sample. Here are pictures of analyzed Sanger data before and after this noise minimization process:

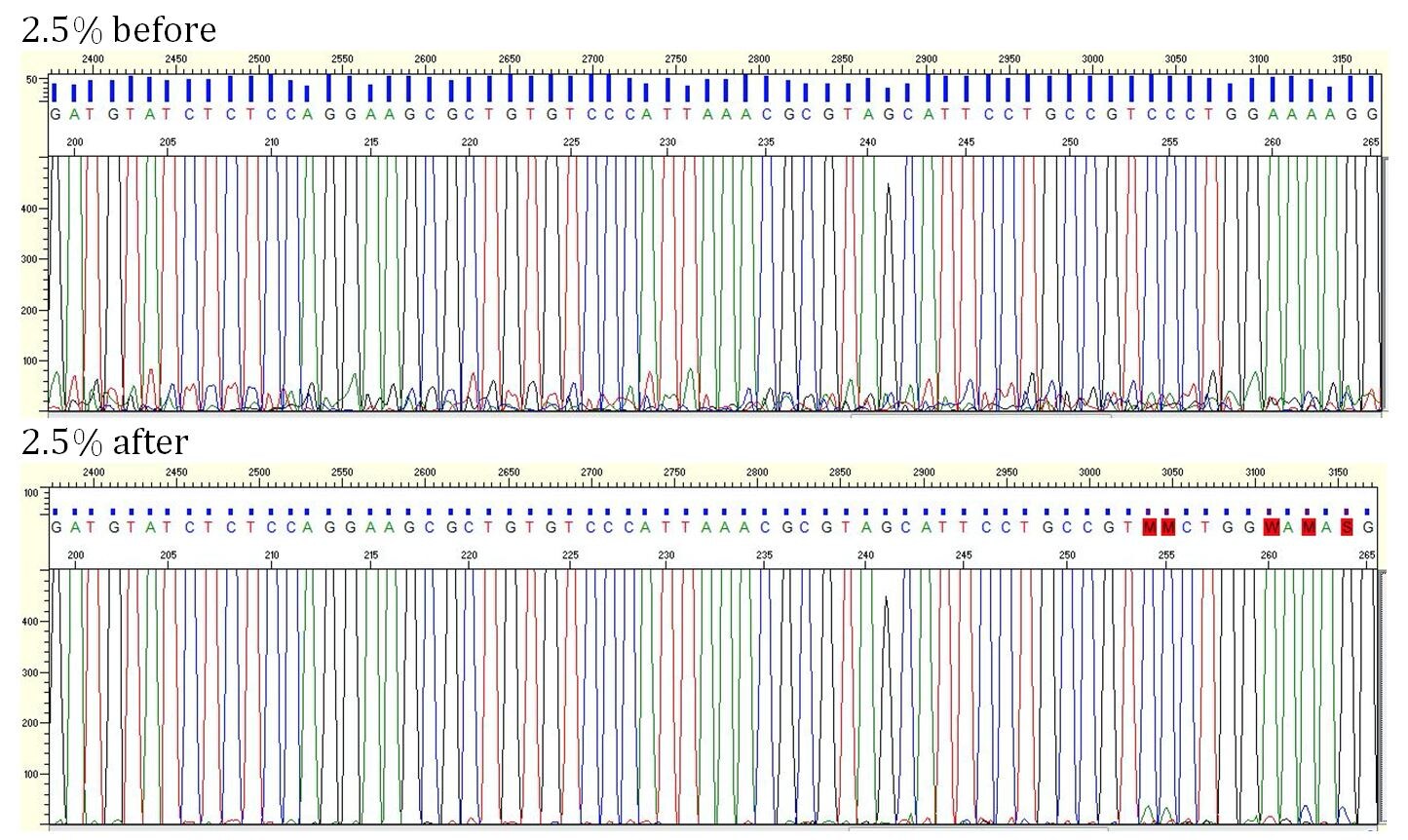

So you want to know how to get these results? Of course you do! Here’s how to breath new life into that old reliable Sanger workhorse of yours:
- Get the new Applied Biosystems Minor Variant Finder (MVF) software.
- Prepare and sequence control and test samples using the same materials and procedures (same primers, same sample clean up method, same instrument, same expert hands, etc.)
- Feed the control forward and reverse reactions and the forward and reverse reactions of the test samples into the MVF software.
- Review the variants called and/or the electropherograms before and after noise minimization.
For a quick overview of the software, check out the video below:
FAQ:
Q1: Do I really have to process control and test samples at the same time on the same instrument?
A1: That’s how you can get the best results. We have not thoroughly investigated sensitivity to system variables to find ways to relax these requirements but we will be doing this.
Q2: Each of my test samples is unique; adding a control doubles cost. Is there any way around this?
A2; Not right now but we are working on ways to possibly eliminate the need for controls. Right now the best strategy is to process all test samples with the same amplicon at the same time.
Q3: What about het-indels?
A3: This is a limitation we are actively working to resolve. MVF relies on comparing forward and reverse reactions to confirm or refute a candidate variant. As you know, this comparison is tricky in the case of het-indels.
Q4: Can the algorithm detect more than one minor variant at the same base position?
A4: Yup.
Q5: Sometimes a sequencing reaction fails in one of the orientations; can I still get results?
A5: Not at the present time. However, providing noise minimized electropherograms and variant detection for data from a single sequencing orientation can be provided in a future release. The quality of the former will not change but, for the latter, high accuracy detection levels depend on forward-reverse confirmation so, without this, false positive rates may increase.
Q6: I run multiple replicates of control samples and/or test samples. Can the software take advantage of these data to provide improved accuracy?
A6: This is a feature planned for a future release.
Q7: My control sample’s primary sequence backbone is not exactly the same as that for the test sample(s), is that a problem?
A7: In general, no. But, at and immediately surrounding base positions where the control and test primary bases differ, noise minimization will not be as effective so there is a possibility of false negatives or positives at those positions. These base positions should be flagged with a red review indicator.
Q8: My control and/or test sample has some germline variants; is that a problem?
A8: If control and test have exactly the same germline variant, it will be taken out by the noise minimization process. If only the test sample has the variant it will be recognized as a variant.
Leave a Reply