Nuclear factor kappa-light-chain-enhancer of activated B cells(NF-κB)、hypoxia-inducible factor-1 alpha(HIF-1α)、およびsignal transducer and activator of transcription(STAT)の3つの転写因子―腫瘍進行を促進する因子―は、炎症反応経路における重要なモジュレーターとして機能します*1 。NF-κBは、炎症反応急性期の調節因子で、効果的な免疫防御および形質転換細胞の排除に必要とされます。更に炎症メディエーターとしてのサイトカインの発現を増加させます。これらのサイトカインには、IL-1、IL-6、TNF-α、リンホトキシン、およびIFN-αが含まれます。さらに、IL-1およびTNF-αはNF-κBの活性化因子であり、フィードバックループを形成します。一方で、多くのさまざまなタイプのがんがNF-κBを恒常的に発現し、腫瘍の形成および進行に関わりをもちます*2 。HIF-1αは、低酸素に対する細胞応答に関与します。酸素レベルが低下すると、HIF-1αは、血管新生および赤血球生成などのプロセスを制御する遺伝子の転写を誘導します。炎症が生じている間、その周囲の免疫細胞において酸素濃度が低下します。この酸素濃度低下は、HIF1-αの発現をより長く持続させるのに寄与します。多くのサイトカイン、例えばIL-1βは、HIF-1αの転写を増加させます。サイトカインの使用は、HIF-1αプロモーターに結合することが示されているNF-κBによって媒介されます*3 。タンパク質のSTATファミリーは、炎症が生じている間、炎症誘導因子のタイプによって複数の機能を果たします。誘導因子のタイプによって、どのサイトカインがトリガーとなるかが決定されます。それらのサイトカインは、STATタンパク質を活性化し、炎症を増加または減少させます。例えば、身体がウイルス感染に遭遇すると、さまざまなIFNがトリガーとなり、抗ウイルス作用を示すSTAT1およびSTAT2を活性化し、炎症を低下させます。それに対し、TAT6はTヘルパー細胞の分化を誘導します。分化タイプに依存して、Tヘルパー細胞は、アレルギー性炎症に正の影響を与え、自己免疫に負の影響を与えます*4。
※本記事の末尾において、腫瘍関連炎症を評価するための抗体ベースのツールについて体系的に学べる無料コンテンツを紹介しています。
NF-κBシグナル伝達経路の概要
NF-κBは、活性化B細胞において免疫グロブリンの軽鎖エンハンサーに結合する転写因子として発見されました。NF-κBファミリーメンバーは、RelA/p65、RelB、c-Rel、NF-κB1、およびNF-κB2の5つが同定されています。NFκ-B1およびNF-κB2(p105およびp100)は、これらの活性型p50およびp52へのタンパク質プロセシングが必要とされます。NF-κBタンパク質は、ホモ二量体またはヘテロ二量体を形成し、それらは通常、抑制性タンパク質のIκBファミリーメンバーに結合します。p50およびp52は転写活性が欠失しており、これらのタンパク質のホモ二量体は翻訳抑制因子として作用するのに対し、p65やRelBなどのファミリーメンバーとヘテロ二量体化すると、活性化転写因子を形成します*5。
がんにおけるNF-κBのシグナル伝達タンパク質としての潜在的な役割は、RELA遺伝子(p65サブユニットをコード)がクローニングされ、ウイルス性v-RELがん遺伝子との相同性が認識された後に明らかとなりました。多くの固形腫瘍およびリンパ性腫瘍においてNF-κBは活性化型ですが、NF-κBカスケード内のタンパク質をコードする遺伝子の大部分は変異していません。ほとんどの場合、がん細胞において、上流のシグナル伝達分子における機能獲得型変異や腫瘍微小環境における成長因子およびサイトカインの分泌の増加に対する反応を経ても、NF-κBは活性化しています*6。
![Phospho-NFκB-p65 [pSer276] Rabbit Polyclonal Antibody](https://www.thermofisher.com/blog/wp-content/uploads/sites/13/2019/07/1-43.jpg)
正常細胞や刺激性シグナルの非存在下では、NF-κBタンパク質(およびプロセシングされていないp105およびp100)は、通常、抑制性タンパク質のIκBファミリーメンバーに結合し、細胞質に局在化します。NF-κB活性化につながる要素は、細胞タイプに依存しますが(図4.1)、可能性のある活性化因子として、パターン認識受容体(Toll様受容体およびNod様受容体)、炎症誘発性サイトカイン(TNF-αおよびIL-1)のための受容体、ならびに抗原受容体が挙げられます。NF-κBの活性化を導くIκBキナーゼ(IKK)は、2つの触媒サブユニットIKKαとIKKβ、および調節サブユニットIKKγ(別名 NEMO)から構成されます。IKKは、IκBをリン酸化し、ユビキチン化および分解を導きます。NF-κBは、抑制性IKKから放出されると、核へ移行し、そこでリン酸化およびアセチル化され、遺伝子転写を活性化します(図4.1)*7。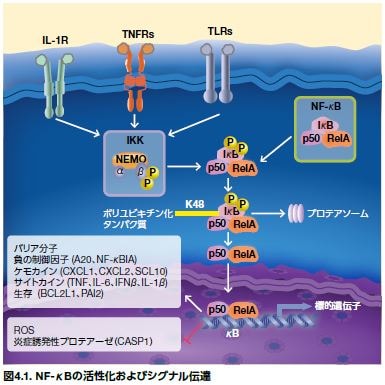
当記事は、腫瘍関連炎症を評価するための抗体ベースのツールガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
NF-κB調節プロセス
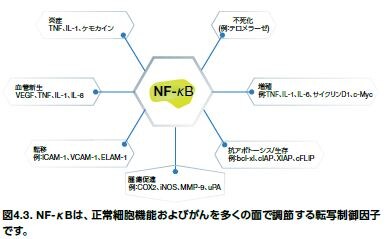
がんにおいて、NF-κBは2つの役割を担っています。急性炎症反応におけるこの転写因子の完全活性化は、がん細胞に向けられた細胞傷害性免疫細胞の活性増加に関連します。しかしながら、NF-κBは、多くのがんの形態において継続的に活性化状態にあり、大量の腫瘍形成反応を誘発します。NF-κB活性化は、アポトーシスを阻害する遺伝子や、細胞生存を促進する遺伝子発現を誘導し、腫瘍微小環境内の酸化ストレスに抵抗します。
NF-κBは、TNF-α、IL-1、IL-6、およびIL-8などの多くのサイトカインの発現にも関与します(図4.2)*5。 腫瘍形成に関与する多くの他の細胞プロセスは、NF-κBによっても制御されています。これらの細胞プロセスには、細胞増殖、細胞接着、上皮間葉転換、細胞浸潤、転移、および血管新生、ならびに他のプロセスが含まれます (図4.2)。NF-κBは、がん幹細胞において活性化されることも示されており、がん細胞の少数の亜集団が腫瘍成長を媒介し、化学療法に対する耐性を持つと考えられています*6。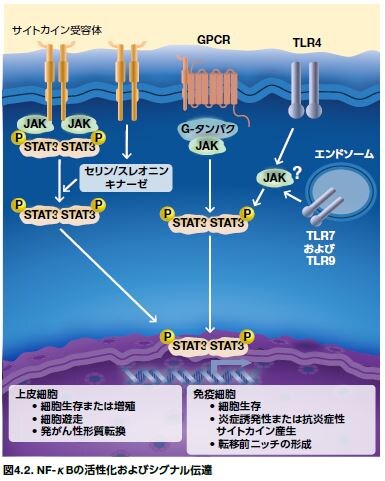
炎症およびSTAT3シグナル伝達
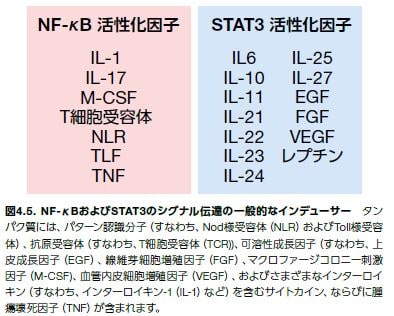
インターフェロンおよびIL-6は、ヤヌスキナーゼ(JAK)-シグナル伝達および転写活性化因子(STAT)シグナル伝達経路の活性化因子として同定された最初のメディエーターです。複数のサイトカインおよび成長因子が、この経路を介するシグナルとして知られています(図4.3および図4.5)*8。7種類のSTATタンパク質ファミリーメンバーのうち、STAT3およびSTAT5が最もがんの発症に関与していると思われます。いくつかの最近の研究において、Toll様受容体(TLR)およびNod様受容体(NLR)は、サイトカインのように、JAK–STAT3経路を活性化し、続いて悪性細胞におけるTLRの発現を促進し、さらに腫瘍進行を促進することが示されています。正常細胞において、STATシグナル伝達は一過性ですが、成長因子およびサイトカインからJAK-STAT3経路までの増強された自己分泌型および傍分泌型のシグナル伝達は、遺伝的不安定性および腫瘍関連炎症に寄与すると考えられます。不活性型のSTATタンパク質は、細胞質に存在します。膜受容体の刺激は、JAKファミリーの受容体関連チロシンキナーゼの活性化を誘導し、それによりSTATがリン酸化されます。JAK1は、STAT3の主要な活性化因子です。リン酸化STATタンパク質は、Src相同領域(SH2)の相互作用を介して、ホモまたはヘテロ二量体を形成します。二量体STATタンパク質は、核に入り、遺伝子転写を制御します(図4.3)*6,10。
NF-κBおよびSTAT3クロストーク
NF-κBおよびSTAT3は、膨大な数のがんの発症に関与する標的遺伝子において、異なりかつ重複する役割を果たしています。STAT3は、3,000以上の異なる遺伝子のプロモーター領域に結合し、NF-κBファミリーはさらに多くのプロモーターに結合している可能性があると推測されています。腫瘍形成およびがん進行の過程で、一部の遺伝子/経路はNF-κBおよびSTAT3の両方の共通の標的となると考えられます。注目すべきは、両方の転写因子が、抗アポトーシスプロセス、血管新生、サイトカイン産生、炎症、サイトカイン遺伝子、細胞周期、および他のプロセスを制御する遺伝子の調節因子であることです*6。
NF-κBおよびSTATは、多種多様なメカニズムを介して、互いに影響し合います(図4.4)。1つのメカニズムは、直接的な物理的結合を介して生じ、転写活性またはDNA結合に影響を与えます。STAT3は、p65およびp50と相互作用することが示されており、それにより、NF-κBのSTATプロモーター領域への局在化やSTAT3のNF-κB結合領域への局在化が促進されます。STAT3の他に、p53、エストロゲン受容体、ATF3、SMAD3、およびSMAD4などの他の転写因子もNF-κBに結合することも知られています。
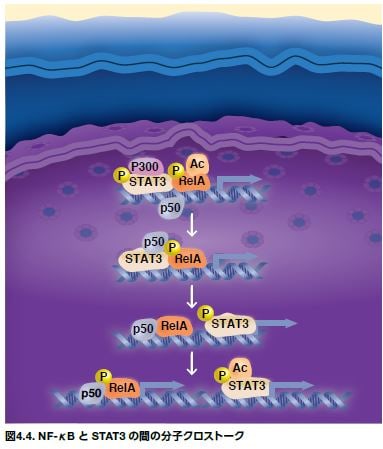
NF-κBおよびSTAT3などの転写因子は、それらのプロモーターまたはエンハンサー配列上のもう一方に近接するDNA配列にも結合し、転写制御機構の必要なコンポーネントをリクルートします。さらに、STAT3はアシルトランスフェラーゼp300をNF-κBにリクルートすることが示されています。RelAの翻訳後アシル化により、核への局在化が増加することは注目に値します。STATを介するNF-κBのアシル化がNF-κB活性の持続を増加させるというフィードフォワード機構が提唱されていますが、腫瘍微小環境における成長因子やサイトカインによる刺激と同様です。これは、がんにおけるNF-κBの恒常的な活性化に寄与します。NF-κBが活性化されると、IL-6などのサイトカインの放出が生じ、それによってSTAT3が活性化されます(図4.5)*5,6。
当記事は、腫瘍関連炎症を評価するための抗体ベースのツールガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
NF-kBおよびSTAT3シグナル伝達のインデューサー
腫瘍微小環境内では、間質細胞、腫瘍浸潤性リンパ球、マクロファージ、および他の細胞タイプが細胞表面分子(TLRおよびNLRなど)を発現するか、NF-κB経路およびSTAT3経路を活性化する可溶性因子を産生しています。当社の抗体検索ツールを使用して、これらならびにNF-κBおよびSTAT3シグナル伝達の他のインデューサーを認識する抗体を見つけてください。一次抗体および二次抗体の膨大なポートフォリオに加えて、当社では、お客様の研究目標の達成のために、幅広い手動および自動のウェスタンブロット装置ならびに試薬を提供しています。
InvitrogenブランドのiBind™およびiBind Flex Western Deviceは、ウェスタンワークフローにおける免疫検出工程を自動化するためのSequential Lateral Flow(SLF)技術を採用
従来のマニュアル処理法では、複数の抗体を準備・交換し、さらに目的ブロットを含むトレイ中で数時間かけて反応します。Invitrogen™iBind™ Western Systemは、免疫検出工程を自動で行う電源・電池不要の装置です。抗体を充填するだけで、3時間後に抗体抗原反応は終わり検出の準備が整います。マニュアルウェスタンブロッティングの改良により誕生したiBind Western Systemは、ハンズオンタイムの短縮およびより一貫性のある結果を可能とします。
- 柔軟性̶同条件でも、異なる条件下でも、一度に最大1枚のMidi Gel、2枚のMini ゲル、または6枚のカットしたゲルの処理が可能
- 適合性̶ニトロセルロース膜またはPVDF膜を使用して、直接標識で一次/二次抗体を検出(AP、HRP、または蛍光標識)
細胞表面タンパク質および細胞内タンパク質の検出には、再現性に優れ、全てのウェスタン検出プロトコルに適合する、当社の自動化されたウェスタン処理システムをご使用ください。
インターロイキン-6(IL-6)は、強力な炎症誘導因子で、さまざまなT細胞サブセット、マクロファージ、好中球、上皮細胞、腫瘍細胞、および腫瘍微小環境内の他の細胞タイプによって分泌されます。IL-6および他のSTAT3誘導因子を検出するためのELISAは、こちらをご覧ください。
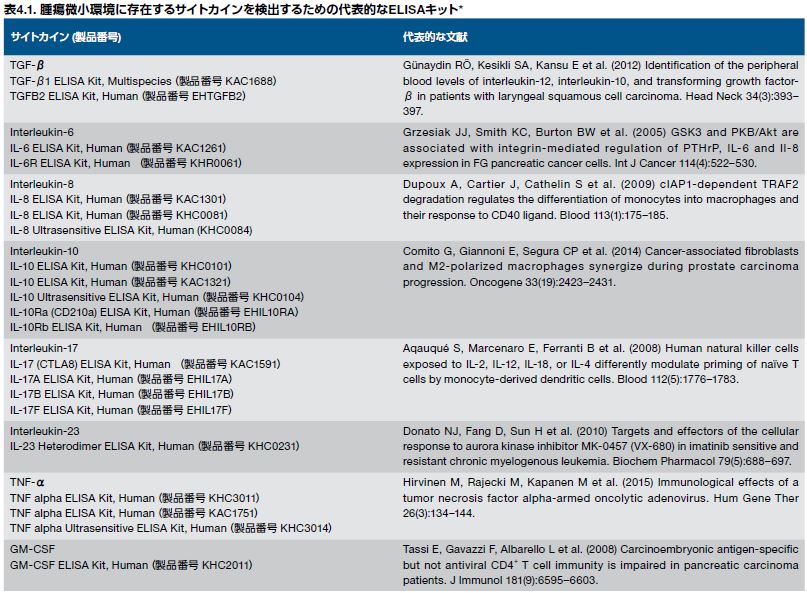 これらおよび他のサイトカインを検出するためのこの他のヒト用キットならびに複数のさまざまな生物種由来のタンパク質を検出するためのキットについてはこちらをご覧ください。
これらおよび他のサイトカインを検出するためのこの他のヒト用キットならびに複数のさまざまな生物種由来のタンパク質を検出するためのキットについてはこちらをご覧ください。
がん標的治療
腫瘍進行の発展においてNF-κBおよびSTAT3の転写因子経路が中心的役割を果たしていると想定し、NF-κBおよびSTAT3のシグナル伝達経路ならびにこれらの経路に関連するいくつかのメディエーターを阻害する薬剤について評価するための数多くの前臨床試験および進行中の臨床試験が存在しています*11,12。 NF-κBを直接的に標的とする薬剤は現在承認されていませんが、NF-κBを間接的に標的とするFDA承認薬や前臨床試験中の化合物は存在します。Bortezomib―IκBαの分解を抑制することによってNF-κBを不活性化するプロテアソーム阻害剤―は、多発性骨髄腫およびマントル細胞リンパ腫の治療薬としてFDAに承認されている他、承認されていませんが、前立腺がん治療薬としても検討されています*13,14。
NF-κBシグナル伝達を標的とする戦略には、以下のアプローチの開発が含まれます
- プロテアソーム阻害剤は、IκBαのユビキチン化および分解に関与します。そのため、プロテアソーム阻害剤は、IκBαの半減期を延ばす可能性があります。
- NF-κB(RelA)の翻訳後修飾を阻害するアセチル化阻害剤は、その核局在化を減少させます。
- 遺伝子導入、細胞透過性ペプチド、およびsiRNAは、がんにおけるNF-κBシグナル伝達の阻害について研究するためのさらなるアプローチです*15。
STAT3シグナル伝達を標的とする戦略には、以下のアプローチの開発が含まれます
STATシグナル伝達の阻害には、数多くの戦略が使用されています。プロテインキナーゼは、幅広く薬理療法のターゲットとされています。STATの阻害では、STATのリン酸化に関与するチロシンキナーゼがターゲットとされます。STATは、リン酸化に続いて、二量化および活性化されます。乳がん細胞におけるチロシンキナーゼ阻害は、培養細胞において、成長を阻害し、恒常的なSTAT3活性化の阻害と関係することが示されています。タンパク質の脱リン酸化も重要な調節シグナルで、シグナルをオフに切り替えたりオンに戻したりします。そのため、タンパク質チロシンホスファターゼの調節は、STATシグナル伝達を阻害するためのもう一つの有望なアプローチとして提案されています。STATシグナル伝達の生理学的タンパク質阻害剤には、STAT活性化を直接的または間接的に制御する内在性タンパク質が含まれます。このようなタンパク質の例として、SH2ドメイン含有タンパク質(サイトカインシグナル伝達抑制因子;SOCS)、JAK結合タンパク質(JAB)、およびSTAT誘導STAT阻害剤(SSI)があります。その他のSTATシグナル伝達の阻害については、二量化の阻害物質、STAT DNA-結合および転写活性の阻害剤、ならびにSTATアンチセンスオリゴヌクレオチドが挙げられます。16 NF-κBシグナル伝達およびがんの進行を制御する分子経路について理解を深めることは、将来の新しい標的治療オプションの拡大に寄与すると考えられます。
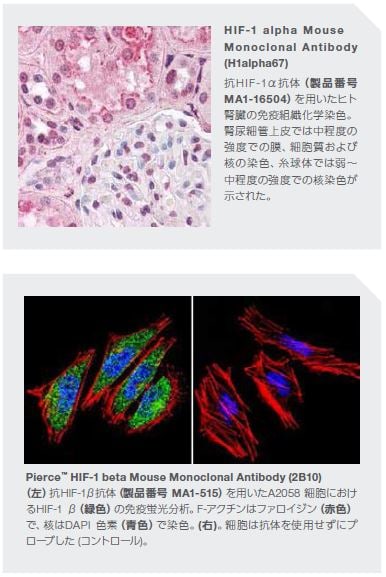
低酸素、炎症、およびがん
正常な生理的条件下の低酸素―すなわち、低い酸素濃度レベル―の状態は、創傷治癒および発生過程に寄与します。しかしながら、無制御な低酸素状態は、虚血性心疾患、炎症性腸疾患、がんおよび他の症状など複数の異なる医学的症状に関連します。腫瘍微小環境内において、低酸素は通常の状態であり、腫瘍形成を促進します。転写制御は、細胞の順応を可能にする多様な一連の遺伝子によって大部分が媒介されています。低酸素に対する細胞応答は、低酸素誘導因子(HIF)として知られる一群のタンパク質によって媒介されます。HIF は、HIF-1α、-2α、および-3αの3つのファミリーメンバーからなり、HIF-1ががん進行に関与するとされています*17,18。 低酸素領域の形成に寄与するいくつかの要因は、慢性炎症組織に関連します。これには、過度の炎症および線維症による微小血管系内酸素流の断絶、炎症が存在する組織細胞および代謝的に活性な浸潤性免疫細胞による酸素消費量の増加などが含まれます*19。
HIF-1 の活性は、以下の観察から実証されているように、炎症と血管新生との関連について明確にされています:結腸直腸腫瘍において、COX-2はHIF-1による低酸素により転写誘導され、腫瘍生存および血管新生が促進されます。急速に成長する腫瘍において、HIF-1 は、アポトーシス抵抗性、血管リモデリングなどの数多くの細胞プロセスの活性化に関与し、腫瘍進行の強力なメディエーターである血管内皮細胞増殖因子(VEGF)などの血管新生に不可欠な数多くの遺伝子を誘導します*20。 VEGF ファミリーメンバーは、血管透過性を増加させ、内皮細胞の増殖を誘導し、白血球粘着能を亢進し、新生血管の血管径を調節します。VEGF ファミリータンパク質と同族受容体の相互作用は、PI3K/ Akt、Ras/Raf-MEK/ Erk、eNOS/NOなどのがん細胞の増殖・生存における全ての重要因子のシグナル伝達経路を活性化します。HIF-1は、VEGF 調節の他、STAT3、NF-κB、mTOR、TGF-βなどのいくつかのさまざまな転写因子の関与するメカニズムを介して発がん促進活性を発揮します。HIF-1が腫瘍形成に関連する制御遺伝子に中心的に関与しているという仮定のもと、HIF-1 シグナル伝達に関与する特定のメディエーターおよびターゲットを阻害する薬剤の開発に多くの力が注がれています。
転写因子の NF-κBおよびSTAT3は炎症反応の重要な調節因子で、がんの発生を促進し、HIF-1αと連携して腫瘍進行を促進します。大部分の新生組織の微小環境における炎症性成分の存在は、血管新生、ホルモンに対する抵抗性(ホルモン依存性腫瘍における)および抗腫瘍免疫を高頻度で増強する結果となっています。腫瘍細胞の生存、増殖、そして最終的な浸潤および転移は、全て腫瘍部位に存在する炎症メディエーターによって制御されています*1,21。
【無料ダウンロード】腫瘍関連炎症を評価するための抗体ベースのツール
当記事は、腫瘍関連炎症を評価するための抗体ベースのツールガイドブックからの抜粋です。以下の内容を含むPDFは下記から無料でダウンロード頂けます。
- 慢性炎症およびがん
- がんドライバー遺伝子および炎症
- 腫瘍微小環境
- シグナル伝達および腫瘍関連炎症
- 要約および付録
【無料ダウンロード】がん研究を促進させる遺伝子解析技術ハンドブック
また、複数のがん研究領域における興味深い最新の発見を可能にする遺伝子解析技術を紹介するハンドブックも配布しています。併せてごらんください。
参考文献:
1. Porta C, Larghi P, Rimoldi M et al. (2009) Cellular and molecular pathways linking inflammation and cancer. Immunobiology 214(9–10):761–777.
2. Medzhitov R (2010) Inflammation 2010: new adventures of an old flame. Cell 140(6): 771–776.
3. Scholz CC, Taylor CT (2013) Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol 13(4):646–653.
4. Kaplan MH (2013) STAT signaling in inflammation. JAKSTAT 2(1):e24198.
5. Hoesel B, Schmid JA (2013) The complexity of NF-kB signaling in inflammation and cancer. Mol Cancer 12:86.
6. Grivennikov S, Karin M (2010) Dangerous liaisons: STAT3 and NF-kB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 21(1):11–19.
7. Israël A (2010) The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb Perspect Biol 2(3):a000158.
8. Yu H, Lee H, Herrmann A, et al. (2014) Revisiting STAT3 signalling in cancer:new and unexpected biological function. Nat Rev Cancer 14(11):736–746.
9. Khiong K, Adhika OA, Chakravitha M (2010) Inflammation, immunity, and cancer: the role of transcription factors NF-kB and STAT3. Maj Kedokt Indon 60(8):369–373.
10. Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9(11):798–809.
11. Furgan M, Akinleye A, Mukhi N et al. (2013) STAT inhibitors for cancer therapy. J Hematol Oncol. 6:90.
12. Miller SC, Huang R, Srilatha S et al. (2010) Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanisms of action. Biochem Pharmacol 79(9):1272–1280.
13. Li C, Chen S, Yue P et al. (2010). Proteasome inhibitor PS-341 (Bortezomib) induces calpain-dependent IkBa degradation. J Biol Chem 285(21):16096–16104.
14. National Cancer Institute. FDA approval for Bortezomab. http://www.cancer.gov/about-cancer/treatment/drugs/fda-bortezomib. Accessed December 2, 2015.
15. de Lartigue J (2013) The inflammation link: NF-κb remains a difficult but intriguing target. OncLive http://www.onclive.com/publications/oncologylive/2013/june-2013/the-inflammation-link-nf-b-remains-a-difficult-butintriguing-target/2. Accessed April 2015.
16. Turkson J, Jove R (2000) STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19(56):6613–6626.
17. Ke Q, Costa M (2006) Hyoxia-inducible factor-1 (HIF-1). Mol Pharmacol 70(5):1469–1480.
18. Semenza GL (2014) Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 9:47–71.
19. Vaupel P, Mayer A (2014) Hypoxia in tumors: pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv Exp Med Biol 812:19–24.
20. Peng XH, Karna P, Cao Z et al. (2006) Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1alpha signal pathways increases resistance to apoptosis by up-regulating survivin gene expression. J Biol Chem 281(36):25903–25914.
21. Balamurugan K (2015) HIF-1 at the crossroads of hypoxia, inflammation and cancer. Int J Cancer DOI:10.1002/ijc.29519.
研究用にのみ使用できます。診断目的およびその手続き上での使用はできません。
記事へのご意見・ご感想お待ちしています

【第2回】エクソソームの濃縮・回収
今回は、さまざまなサンプルからのエクソソ...
Read More
ライブラリー調製の自動化でNGSの可能性を広げる―Ion Chefシステムが変える研究室のワークフロー
前処理の自動化が、NGSの新しい標準をつくる...
Read More
ラボボトルの耐薬品性と気密性とは?~ 意外と知らない基本の話~
ボトル選び、なんとなくで大丈夫? プラス�...
Read More
大腸菌の飼い方~実験に必要な設備をそろえよう
分子生物学の黎明期を支えてきた大腸菌(Esc...
Read More