 Phosphorylation is one of the most common post-translation modifications (PTMs) of proteins; it has been implicated in the pathogenesis of many disease states, including cancer. Phosphorylation requires action by specific kinase enzymes, whose roles have been studied extensively. On the flip side of this PTM stand the phosphatase enzymes, which remove added residues. Much less is known about these enzymes and the circumstances under which dephosphorylation affects signaling pathways. Recent studies show, however, that phosphatases might be potential therapeutic targets for management of tumors.
Phosphorylation is one of the most common post-translation modifications (PTMs) of proteins; it has been implicated in the pathogenesis of many disease states, including cancer. Phosphorylation requires action by specific kinase enzymes, whose roles have been studied extensively. On the flip side of this PTM stand the phosphatase enzymes, which remove added residues. Much less is known about these enzymes and the circumstances under which dephosphorylation affects signaling pathways. Recent studies show, however, that phosphatases might be potential therapeutic targets for management of tumors.
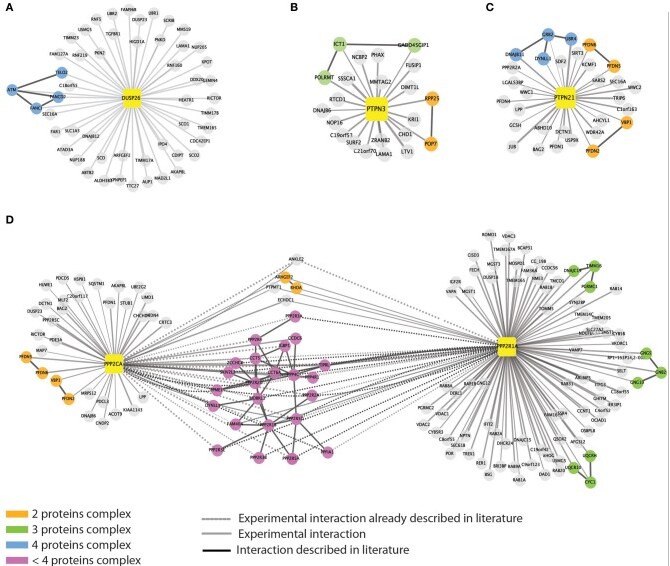
Sacco et al. (2014) took a novel approach to investigating phosphatase dynamics, using affinity proteomics to extract and characterize the protein scaffold involved in presenting the enzyme to its substrate.1 They used network and functional analysis to circumvent the lack of specificity shown by phosphatases for substrate and created a protein interaction network to highlight potential substrates and interactors.
The researchers chose to examine eight phosphatases, identified from previous functional siRNA screening, that modulate the RAS-PI3K pathway. Using transient transfection of tagged proteins in human epithelial carcinoma HeLa cells in conjunction with SILAC (stable isotope labeling with amino acids in cell culture), they first captured the protein complexes using a streptavidin-conjugated avidin beads affinity purification step. The research team then purified the protein complexes using SDS-PAGE and in-gel trypsin digestion before proteomic analysis using an LTQ Orbitrap Velos hybrid ion trap-Orbitrap mass spectrometer (Thermo Scientific). They then identified proteins by searching the data against the human-specific IPI database (version 3.52). The team also confirmed protein identifications via immunoprecipitation and subsequent Western immunoblotting.
Sacco et al. found that the affinity proteomics data identified a number of protein interactors that co-enriched with the phosphatase enzymes under examination, with around 10% of the data reported elsewhere in research literature. Their new affinity proteomics approach also identified new phosphatase interactions such as DLC1 and ATM.
The research team also studied dynamic modulation of the phosphatase interaction network, using TNFα to stimulate the cells after transfection. They found that although the majority of interaction proteins were unaffected, some indeed revealed up- or down-regulation with treatment.
Next, the scientists looked into pathways and signaling networks potentially affected by the phosphatase interactome data they had acquired. Using a KEGG-pathway enrichment analysis tool (DAVID software), they examined the interactor complexes in order to assemble a protein interaction network for the phosphatase enzymes. They used their analyses to construct an interconnected, human phosphatase interactome, which they then used in conjunction with data derived from relevant literature and the mentha PPI database to suggest novel enzyme substrates. The team further tested one of their proposals using in vitro cell culture and co-immunoprecipitation to confirm protein associations and regulation.
The researchers conclude that their approach is a valid method, combining functional analysis with protein identification data to characterize the phosphatase interactome, thus highlighting potential targets for regulation.
Reference
1. Sacco, F., et al. (2014, May) “Combining affinity proteomics and network context to identify new phosphatase substrates and adapters in growth pathways,” Frontiers in Genetics | Systems Biology, 5 (p. 115), doi: 10.3389/fgene.2014.00115.
Post Author: Amanda Maxwell. Mixed media artist; blogger and social media communicator; clinical scientist and writer.
A digital space explorer, engaging readers by translating complex theories and subjects creatively into everyday language.

Acetylation of the Malaria Parasite
Lysine acetylation is a ubiquitous post-translational modifi...
Read More
Holocaust Exposure and Intergenerational Impact via FKBP5 Methylation
Scientists know that, in animal models, stress can produce e...
Read More
C-Terminomics: What’s Happening at the End of the Rainbow
Structurally speaking, all proteins have a beginning and an ...
Read More
New Roles for Lysine Demethylase KDM4A
Lysine methyltransferases and lysine demethylases are two ex...
Read More
Leave a Reply