 Protein levels from vials of whole blood are generally clinically determined using single-protein immunoassays. However, according to research by Geyer et al.,1 a single drop of blood and a mass spectrometer may be all that’s needed to assess human health and disease.
Protein levels from vials of whole blood are generally clinically determined using single-protein immunoassays. However, according to research by Geyer et al.,1 a single drop of blood and a mass spectrometer may be all that’s needed to assess human health and disease.
Currently, single-protein immunoassays are used in clinical practice. However, immunoassays have fundamental limitations when it comes to multiplexing, their specificity for protein isoforms and their incompatibility with hypothesis-free investigations.
Mass spectrometry (MS)-based proteomics is a technology that could tackle these limitations. Another benefit is that the technology could be capable of discovering biomarkers in just a single drop of blood.
High-throughput, quantitative MS-based proteomics approaches are thought to be advantageous. But they are challenging for various reasons: there is a high dynamic range of protein abundances as well as a lack of reproducible, robust and high-throughput proteomic workflows to identify potential biomarkers in large cohorts. In fact, less than 1.5 novel biomarkers per year were established between 1995 and 2010.1
Geyer et al. show how this is now possible via a rapid and robust “plasma proteome profiling” pipeline, developed thanks to vast improvements in MS-based proteomics technologies in recent years. The workflow uses a single-run shotgun approach, does not require protein depletion, and enables quantitative analysis of hundreds of plasma proteomes from only 1 μl of blood, using 20-minute gradients.
This new approach is capable of quantitatively measuring hundreds of plasma proteomes, including members of the inflammatory marker family such as C-reactive protein, as well as the apolipoprotein family, gender-related proteins and more than 40 FDA-approved biomarkers.
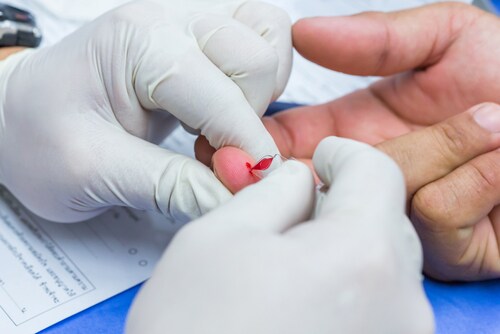
The researchers collected small quantities of blood from 10 individuals, via simple finger pricks, and prepared the samples using protein digestion and the in-StageTip method.2 All preparation steps were performed in a single vial.
Samples were measured using liquid chromatography (LC)-MS consisting of an EASY-nLC 1000 ultra-high-pressure system coupled via a nano-electrospray ion source to a Q Exactive HF Orbitrap mass spectrometer (all Thermo Scientific). MaxQuant was then used for quantitative label-free analysis of the LC-MS/MS data.
Following MS analysis, the team performed data analysis by searching against two different databases: the human UniProt FASTA database and a common contaminants database by the Andromeda search engine. Of the 347 protein groups identified in the 20-minute gradients, 285 were detected in all 10 individuals.
The entire workflow was shown to be robust and highly reproducible and, including sample preparation and data analysis, took less than three hours, highlighting the value of quantifying hundreds of proteins in a very short analysis time. Despite using extremely short measurements, accuracy and precision of the label-free workflow were excellent, with intra-assay correlation of about R2 = 0.98 and coefficients of variation smaller than 20% for the majority of quantified proteins. Further, the ability to use small sample amounts makes blood testing much less invasive, improves cost-efficiency and is clinically attractive.
As part of the same study, to investigate a “deeper plasma proteome,” the team also used a combination of peptide prefractionation, a matching library consisting of depleted plasma, and 100-minute high-performance LC (HPLC) gradients to obtain a ~1,000 protein quantitative proteome. Unexpectedly, the deep plasma proteome contained only 14 additional FDA-approved biomarkers compared with the 49 already found in the 20-minute gradients.
This research shows that plasma proteome profiling from a minuscule amount of blood can deliver an informative portrait of a patient’s health state. Geyer et al. envision its large-scale use in biomedicine.
References
1. Geyer, P.E., et al. (2016) “Plasma proteome profiling to assess human health and disease,” Cell Systems, 2(3) (pp.185–195). doi: http://dx.doi.org/10.1016/j.cels.2016.02.015.
2. Kulak, N.A., et al. (2014) “Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells,” Nature Methods, 11 (pp. 319–324). doi: 10.1038/nmeth.2834.
Post Author: Kathryn Loydall. Kathryn is a science and medical writer with a background in protein chemistry, biochemistry and applied biology. She enjoys making science accessible to a lay audience through writing, illustrations and media. Originally from the UK, she moved to Vancouver Island in 2008 to complete her PhD focussed on sepsis research and x-ray crystallography of monoclonal antibodies. In 2012, Kathryn left the lab behind to start freelancing as a science and medical writer and editor, and hasn’t looked back since.

Analysis of Ketosteroids by Girard P Derivatization
Dehydroepiandrosterone (DHEA); 4-androstene-3,17-dione (AD);...
Read More
Profiling Human Islet Cells with Laser Capture Microdissection
Composed mainly of β-cells, human islet cells secrete h...
Read More
Intact Proteins in Native Conditions with Quadrupole-Orbitrap Mass Spectrometry
Successful research focusing on antibody and drug interactio...
Read More
Autophagy Helps Control Endothelial Permeability
A research team in Glasgow has identified autophagy as a str...
Read More
Leave a Reply