Search
Is RNA Amplification Necessary for Microarrays?
RNA amplification using the Van Gelder and Eberwine technique (1) has been used extensively for target synthesis and labeling in array analysis experiments. Amplification of RNA is clearly necessary when working with limited samples (needle biopsies, LCM samples, etc.), but is it necessary when the availability of sample is not limiting? Is there additional biologically relevant information that can be obtained from array analysis using amplified RNA as a target? In this article we provide a brief synopsis of the findings of Polacek et al. (2) and Feldman et al. (3) on why amplified RNA (aRNA) might be a better choice than unamplified RNA for microarrays.
The Polacek Experiments
Polacek's group tested the fidelity of differential gene expression data using amplified RNA in a well established in vitro model of cytokine [tumor necrosis factor (TNF-α)]-stimulated human aortic endothelial cells (HAEC). 100 ng of total RNA, isolated from HAECs using a glass filter based kit, was amplified using Ambion's
MessageAmp™ aRNA Kit, producing 4-5 µg of aRNA (cRNA). 2 µg of this aRNA was labeled with 33P by reverse transcription using hexanucleotide random primers; 10 µg of Total RNA (unamplified) was labeled with 33P by reverse transcription using Oligo d(T)15 primers. These targets were hybridized to arrays containing 13,284 human cDNA elements.
Analysis of differential gene expression in the cells treated with TNF-α compared to untreated control cells identified 1296 genes in the amplified samples and 155 genes in the unamplified samples whose expression was affected by cytokine treatment. By comparing genes identified with probe prepared by both amplified and unamplified RNA, three categories were created: "genes common in amplified and unamplified RNA", "genes unique to amplified RNA", and "genes unique to unamplified RNA" (Figure 1). Real-time quantitative RT-PCR (qRT-PCR) analysis using SYBR® Green detection of expression differences of a subset of the genes from each category was carried out. A summary of the observed results from qRT-PCR analysis and array analysis is shown in Table 1. The following conclusions were drawn from these validation studies:
(A) Because genes were identified as having differential expression with both preparations of labeled target, "common identified genes" are considered to have the highest probability of being verifiably regulated by TNF-α. In general, the ratio of gene expression with and without TNF-α treatment from amplified RNA had a closer correlation with the qRT-PCR data than ratios from unamplified RNA. 94% of the "common identified genes" show similar kinase-regulation patterns between amplified and unamplified samples.
(B) "Genes unique to amplified RNA" were ranked by P-values, and 24 genes (2.1% of the total 1,150) were selected from the entire range of P-values for validation by qRT-PCR. Similar regulation patterns between array and qRT-PCR analysis were observed in 16 of the 24 genes tested.
(C) Six from the category of "genes unique to unamplified RNA" were validated by qRT-PCR. Of these, three genes showed a similar expression ratio by qRT-PCR analysis.
Thus amplification improved the sensitivity of the array analysis, enabling the identification of biologically regulated genes that might have been missed if unamplified RNA had been used.
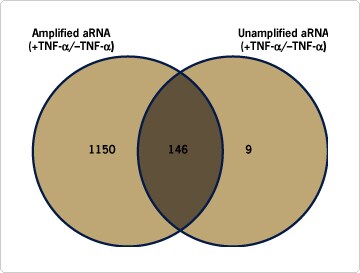
Figure 1. Venn Diagrams Showing the Differential Distribution of Genes Expressed With and Without TNF-α Treatment. Of the total 13,284 genes tested, 1,296 were identified from amplified RNA vs. 146 genes that are commonly represented in both samples.
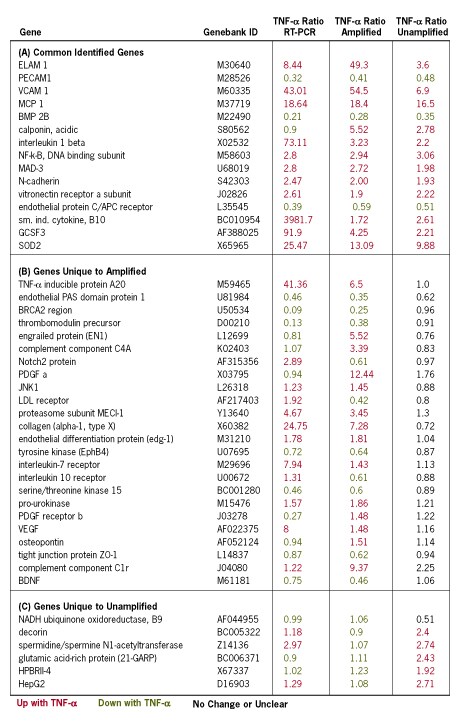
Table 1. Microarray Results. This study involved the comparison of multiple replicate arrays for each condition tested: (+TNF-α and TNF-α) with use of four unamplified and five amplified RNA samples. The gene expression ratios between microarray and quantitative RT-PCR data is presented in this table.
Analysis of differential gene expression in the cells treated with TNF-α compared to untreated control cells identified 1296 genes in the amplified samples and 155 genes in the unamplified samples whose expression was affected by cytokine treatment. By comparing genes identified with probe prepared by both amplified and unamplified RNA, three categories were created: "genes common in amplified and unamplified RNA", "genes unique to amplified RNA", and "genes unique to unamplified RNA" (Figure 1). Real-time quantitative RT-PCR (qRT-PCR) analysis using SYBR® Green detection of expression differences of a subset of the genes from each category was carried out. A summary of the observed results from qRT-PCR analysis and array analysis is shown in Table 1. The following conclusions were drawn from these validation studies:
(A) Because genes were identified as having differential expression with both preparations of labeled target, "common identified genes" are considered to have the highest probability of being verifiably regulated by TNF-α. In general, the ratio of gene expression with and without TNF-α treatment from amplified RNA had a closer correlation with the qRT-PCR data than ratios from unamplified RNA. 94% of the "common identified genes" show similar kinase-regulation patterns between amplified and unamplified samples.
(B) "Genes unique to amplified RNA" were ranked by P-values, and 24 genes (2.1% of the total 1,150) were selected from the entire range of P-values for validation by qRT-PCR. Similar regulation patterns between array and qRT-PCR analysis were observed in 16 of the 24 genes tested.
(C) Six from the category of "genes unique to unamplified RNA" were validated by qRT-PCR. Of these, three genes showed a similar expression ratio by qRT-PCR analysis.
Thus amplification improved the sensitivity of the array analysis, enabling the identification of biologically regulated genes that might have been missed if unamplified RNA had been used.
Figure 1. Venn Diagrams Showing the Differential Distribution of Genes Expressed With and Without TNF-α Treatment. Of the total 13,284 genes tested, 1,296 were identified from amplified RNA vs. 146 genes that are commonly represented in both samples.
Table 1. Microarray Results. This study involved the comparison of multiple replicate arrays for each condition tested: (+TNF-α and TNF-α) with use of four unamplified and five amplified RNA samples. The gene expression ratios between microarray and quantitative RT-PCR data is presented in this table.
The Feldman Experiments
Feldman et al. evaluated the fidelity of microarrays probed with amplified vs. total RNA by comparing microarray data with real-time qRT-PCR. They compared gene expression in two murine tumor cell lines (B16F10 and MC38) using mouse microarrays containing 2601 elements. In their experiments, all array hybridizations were repeated using reciprocal fluorescence to minimize the effects of labeling bias (i.e. each array was hybridized with with Cy™3 labeled B16F10 RNA and Cy5 labeled MC38 RNA, and then a second identical array was hybridized with Cy5 labeled B16F10 RNA and Cy3 labeled MC38 RNA).
The following differences were observed between total RNA and amplified RNA samples:
(A) Correlation coefficients between reciprocal arrays were consistently higher with probes generated by a single or two rounds of amplification than with probes from unamplified RNA.
(B) Total RNA samples showed a higher correlation between duplicate forward or reciprocal arrays than between forward and reciprocal arrays. This was probably partially due to labeling bias. Total RNA also showed increased variability between duplicate arrays compared to amplified RNA. The authors speculate that data generated with total RNA (unamplified) may be more subject to the effects of background on the low-copy number transcripts than amplified RNA.
(C) The correlation between arrays using total RNA and amplified RNA was found to be similar to duplicate arrays probed with total RNA. This observation corroborates the finding that amplification does not significantly bias the gene expression data.
The authors further evaluated whether the individual genes that were differentially expressed in amplified samples would have been identified as such using total RNA. For this experiment, the number of "outliers" defined by authors as "A gene with expression ratios of greater than or equal to 2.0 or less than or equal to 0.5 in two reciprocal microarray analysis" identified using total RNA or amplified RNA (one or two rounds of amplification) were compared. The amplified samples identified approximately 80% of the genes selected as outliers with total RNA, with an 80% concordance between target made by one and two rounds of amplification. Thus amplification yielded approximately 2 times more outliers than total RNA, with 80% of these showing similar trends in expression between amplified and total RNA samples. Validation of the "outliers" between total RNA and amplified RNA was carried out using real-time PCR analysis. The PCR data suggested that the majority of outliers identified using amplified RNA are valid. Thus Feldman et al. also conclude that all RNA samples should undergo at least one round of amplification to maximize the quality of array data for detection of differentially expressed genes.
SYBR Green is a registered trademark of Molecular Probes, Inc.
Cy is a trademark of Amersham Biosciences.
The following differences were observed between total RNA and amplified RNA samples:
(A) Correlation coefficients between reciprocal arrays were consistently higher with probes generated by a single or two rounds of amplification than with probes from unamplified RNA.
(B) Total RNA samples showed a higher correlation between duplicate forward or reciprocal arrays than between forward and reciprocal arrays. This was probably partially due to labeling bias. Total RNA also showed increased variability between duplicate arrays compared to amplified RNA. The authors speculate that data generated with total RNA (unamplified) may be more subject to the effects of background on the low-copy number transcripts than amplified RNA.
(C) The correlation between arrays using total RNA and amplified RNA was found to be similar to duplicate arrays probed with total RNA. This observation corroborates the finding that amplification does not significantly bias the gene expression data.
The authors further evaluated whether the individual genes that were differentially expressed in amplified samples would have been identified as such using total RNA. For this experiment, the number of "outliers" defined by authors as "A gene with expression ratios of greater than or equal to 2.0 or less than or equal to 0.5 in two reciprocal microarray analysis" identified using total RNA or amplified RNA (one or two rounds of amplification) were compared. The amplified samples identified approximately 80% of the genes selected as outliers with total RNA, with an 80% concordance between target made by one and two rounds of amplification. Thus amplification yielded approximately 2 times more outliers than total RNA, with 80% of these showing similar trends in expression between amplified and total RNA samples. Validation of the "outliers" between total RNA and amplified RNA was carried out using real-time PCR analysis. The PCR data suggested that the majority of outliers identified using amplified RNA are valid. Thus Feldman et al. also conclude that all RNA samples should undergo at least one round of amplification to maximize the quality of array data for detection of differentially expressed genes.
SYBR Green is a registered trademark of Molecular Probes, Inc.
Cy is a trademark of Amersham Biosciences.
References
- Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH (1990) Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA 87: 1663-67.
- Polacek DC, Passerini AG, Shi C, Francesco NM, Manduchi E, Grant GR, Powell S, Bischof H, Winkler H, Stoeckert CJ Jr, Davies PF. (2003) Fidelity of enhanced sensitivity of differential tanscription profiles following linear amplification of nanogram amounts of endothelial mRNA. Physiol Genomics 13:147-56.
- Feldman AJ, Costouros NG, Wang E, Qian M, Marincola FM, Alexander HR, Libutti SK (2002) Advantages of mRNA amplification for microarray analysis. Biotechniques 33(4): 906-14.