Search
SNP Genotyping Support—Troubleshooting
Find valuable information.
Optimize your experiments to get the best results. We’ve compiled a detailed knowledgebase of the top tips and tricks to meet your research needs.
View the relevant questions below:
Beginning your experiment?
Visit our
General
This could happen if the assay fails functional testing. All human SNP genotyping assays get tested with a panel of human gDNA samples. The assay must show amplification with at least one cluster in order to pass. When there is no amplification you will be notified of the failure and not charged for the assay. The assay may have failed for the following reasons:
-Input sequence was incorrect (cDNA instead of gDNA)
-Input sequence was not human
-Input sequence was not appropriately pre-screened
-Signal from NTC was less than 0.5 units from samples (assay did not give high fluorescence signal)
To resolve this, please consult the guidelines in the Design and Ordering Guide to properly prepare the sequence for the Custom Assay Design Tool. If the sequence was not human, make sure to select non-human at the species filter step. Nonhuman assays do not get functionally tested.
There are several reasons for a SNP assay not amplifying, including:
-DNA may not be accurately quantitated
-Degraded DNA
-Inhibitors in the sample
-Error in reaction setup
Check out our troubleshooting tool for more details.
If you are not getting calls in the instrument software, you can try the free TaqMan® Genotyper™ Software. This program has an improved algorithm which allows it to make calls that are often missed by the SDS software. See the example below.
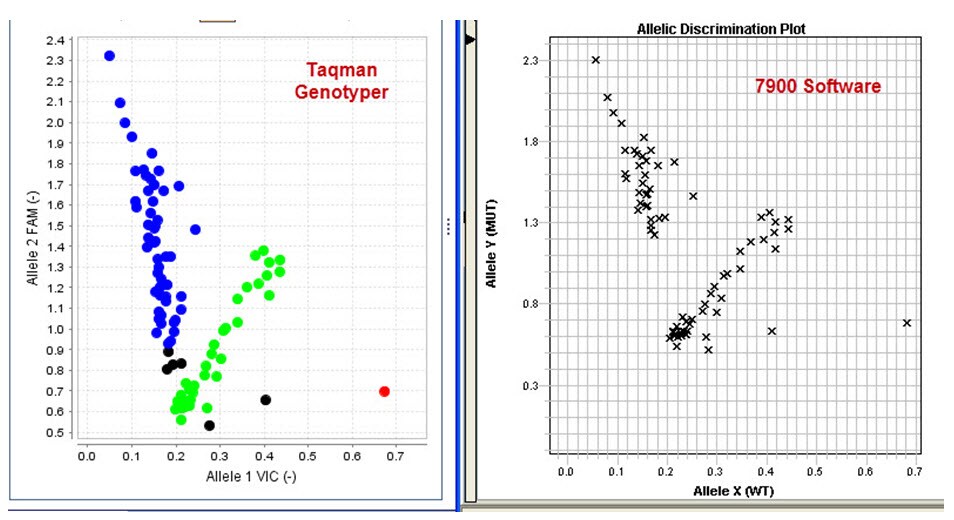
The same data was analyzed with either the TaqMan® Genotyper™ Software or the standard autocalling with the instrument (7900) software. While the data has a problem with trailing clusters, there are clearly 3 clusters present. The TaqMan® Genotyper™ Software is able to call the majority of the samples, while the autocalling cannot.
Data Analysis and TaqMan® Genotyper™ Software
Depending on the assay, you may want to try either reducing or increasing the number of cycles. Our newer instrument software can even allow you to view the traces if you collect the real-time data on the instrument.

Check the Minor Allele Frequency (MAF) of the SNP. You may need a larger sample size in order to see the allele. You can use the Hardy-Weinberg equation to determine if the minor allele is detectable in your sample size or not. Follow the example here (p. 4-2).
Multiple clusters such as in the example below could be due to a hidden SNP under the probe or primer. Search dbSNP for other SNPs around the target SNP. If the nontarget SNP has a low MAF it will usually not be a problem. If the nontarget SNP is under a primer, try to redesign and mask it as an “N”. Another possibility is that the region is within a copy number variation, in which case you will have to evaluate with a TaqMan® Copy Number Assay as well.
I have trailing clusters in my allelic discrimination plot. What can I do to improve the clustering?
Trailing clusters are often due to variation in gDNA quality or concentration. Please see the example here for more details.

The software does not allow assay IDs or sample IDs to be modified. If a typing error occurred or an edit is required, this change must be made in the original experiment file. The software does, however, allow you to create your own assay name and add additional information related to an assay or sample through Setup --> Assays or Setup --> Samples.
An assay or sample may be deleted from a study only if there is no data or wells associated with it. Upon import of an experiment, the software collects all the assays and samples from the plate and lists them in the Setup > Assays or Setup > Samples workspaces. The assays and samples are stored in these workspaces as a library, and remain there even if you delete the experiment from the Study. Deleting the experiment will remove any data (wells) associated with the assays or samples, but not the assays or samples from the library. The assays and samples must then be deleted from these workspaces to remove them from the Study.
- Go to the Start button > Programs > TaqMan® Genotyper Software
- Right-click on the program and choose “Run as Administrator”
- If that does not work, go back to the same menu and choose “Properties”
- Choose the “Compatibility” tab, and check “Run this program as administrator”
- Click “Apply”
- You may have to restart the computer for the settings to apply

Bookmarking is a unique feature in TaqMan® Genotyper™ Software that allows you to tag a data point or well while reviewing results in a Study. For example, in reviewing a cluster plot for an assay, a data point is observed to be somewhat between clusters. You can set a bookmark for this data point to denote this well for further investigation. The bookmark persists between the Results workspace and Quality Control workspace, so you can easily identify the data point in a cluster plot, experiment plate view, or on the samples tab. Bookmarks are cleared upon exit from a Study or exit from the application.
The polymorphism sequence info can be entered into the software through Setup >Assays. You can import an assay information file (AIF) that contains this info for your assays (AIFs are shipped with assay orders), or manually enter this info for each assay using the edit assay feature. The polymorphism sequence info will be displayed in the assays table under allele1 base and allele2 base, in the results table in the calls column, in the cluster plot display in the x-axis and y-axis titles, and in the export files as genotypes. If no sequence information is entered for an assay, the default display for genotype calls will use the dye names, such as VIC®/VIC®, VIC®/FAM™ or FAM™/FAM™ dyes.
A reference panel is helpful in large studies to mark your reference samples. Please follow the directions here on how to set up a reference panel.
For Research Use Only. Not for use in diagnostic procedures.