Search
Vector-Based RNAi Support – Getting Started
Find valuable information.
Optimize your experiments to get the best results. We’ve compiled a detailed knowledge base of the top tips and tricks to meet your research needs.
View the relevant questions below:
Having problems with your experiment? Visit our
siRNA Vectors
Vector technologies allow you to:
- Achieve transient or stable target knockdown
- Perform RNAi in any cell type, even hard-to-transfect, primary, and non-dividing cells
- Regulate gene inhibition with inducible siRNA expression
- Select for a pure population of cells stably expressing an siRNA sequence
- Control gene expression in vivo with tissue-specific promoters
Please review these tips for cloning siRNA templates into pSilencer™ vectors.
Please visit our Transfection Support Center to find technical resources, tips and tricks, and troubleshooting information on transfection.
shRNA RNAi Vectors
For efficient shRNA expression, a Pol III type promoter is used. These Pol III promoters contain all of their essential elements upstream of the expressed RNA and terminate with a short polythymidine tract. Once the shRNA is expressed, it is transported from the nucleus and processed into siRNA in the cytoplasm by the enzyme Dicer. Dicer preferentially recognizes shRNAs generated from a Pol III promoter because they carry no 5’ or 3’ flanking sequences. The siRNAs enter into RISC complexes and generate an RNAi response in mammalian cells.
Short hairpin RNA (shRNA) is an artificially designed class of RNA molecules that can trigger gene silencing through interaction with cellular components common to the RNAi and miRNA pathways. Although shRNA is a structurally simplified form of miRNA, these RNA molecules behave similarly to siRNA in that they trigger the RNAi response by inducing cleavage and degradation of target transcripts (Brummelkamp et al., 2002; Paddison et al., 2002; Paul et al., 2002; Sui et al., 2002; Yu et al., 2002). An RNA Polymerase III (Pol III), such as U6 and H1, drives transcription of shRNA transcripts. shRNA hairpins are exported from the nucleus and processed by Dicer into the cytosol, resulting in siRNA.
Exogenous short hairpin RNAs can be transcribed by RNA Polymerase III (Paule and White, 2000) and generally contain the following structural features:
- A short nucleotide sequence ranging from 19–29 nucleotides derived from the target gene, followed by
- A short spacer of 4–15 nucleotides (i.e., loop) and
- A 19–29 nucleotide sequence that is the reverse complement of the initial target sequence.
The resulting RNA molecule forms an intramolecular stem-loop structure that is then processed to an siRNA duplex by the Dicer enzyme.
We offer our pENTR™/U6 (Cat. No. K492000) and pENTR™/H1/TO (Cat. No. K494500) vectors for shRNA delivery. Both vectors are Gateway® compatible and drive expression through either the U6 or H1/TO promoter, respectively. The pENTR™/H1/TO vector is for inducible shRNA expression, while the pENTR™/U6 can be used for constitutive expression. If you want to design shRNA oligos compatible with both vectors, select the pENTER™/U6 vector. Please see the image below for the vector maps:
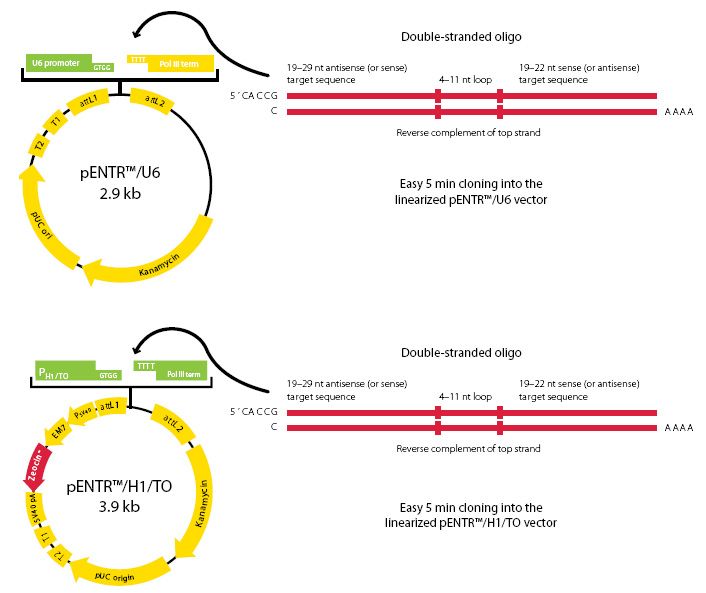
The BLOCK-iT™ Inducible H1 and U6 Entry Vector Kits use either the Pol III–dependent H1 or the U6 promoter, respectively. The H1 promoter is modified to contain two flanking tetracycline operator (TetO2) sites within the H1 promoter. This allows the shRNA expressed from this promoter to be regulated in cells that express the tetracycline repressor (TR) protein. Both the H1 and the U6 are Pol III type promoters; however, there may be some minor differences in their effectiveness, depending on the cell line used.
TO stands for tetracycline operator, as this entry vector contains elements required for tetracycline-inducible expression of the shRNA in mammalian cells. The presence of the Tet operator sequences enables the shRNA of interest to be expressed in a tetracycline-dependent manner, thereby making this an inducible system.
You will need a double-stranded oligo that encodes the shRNA of interest to be cloned into one of the above-mentioned vectors. Use our RNAi Designer to design and synthesize two complementary single-stranded DNA oligonucleotides, with one encoding the shRNA of interest.
You will want to anneal equal amounts of the top- and bottom-strand oligos to generate the ds oligos. If your single-stranded oligos are supplied lyophilized, resuspend them in water or TE buffer to a final concentration of 200 µM before use. We generally perform the annealing reaction at a final single-stranded oligo concentration of 50 μM. Annealing at concentrations lower than 50 μM can significantly reduce the efficiency. Note that the annealing step is not 100% efficient; approximately half of the single-stranded oligos remain unannealed even at a concentration of 50 μM. Please see the steps below:
1. In a 0.5 mL sterile microcentrifuge tube, set up the following annealing reaction at room temperature.
Reagent Amount
“Top-strand” DNA oligo (200 μM) 5 μL
“Bottom-strand” DNA oligo (200 μM) 5 μL
10X Oligo Annealing Buffer 2 μL
DNase/RNase-Free Water 8 µL
Total volume 20 μL
2. If reannealing the lacZ ds control oligo, centrifuge its tube briefly (~5 seconds), then transfer the contents to a separate 0.5 mL sterile microcentrifuge tube.
3. Incubate the reaction at 95°C for 4 minutes.
4. Remove the tube containing the annealing reaction from the water bath or the heat block, and set it on your laboratory bench.
5. Allow the reaction mixture to cool to room temperature for 5–10 minutes. The single-stranded oligos will anneal during this time.
6. Place the sample in a microcentrifuge and centrifuge briefly (~5 seconds). Mix gently.
7. Remove 1 μL of the annealing mixture and dilute the ds oligo as directed.
8. Store the remainder of the 50 μM ds oligo mixture at –20°C.
You can verify the integrity of your annealed ds oligo by agarose gel electrophoresis, if desired.
We suggest running an aliquot of the annealed ds oligo (5 μL of the 500 nM stock) and comparing it to an aliquot of each starting single-stranded oligo (dilute the 200 μM stock 400-fold to 500 nM; use 5 μL for gel analysis). Be sure to include an appropriate molecular weight standard. We generally use the following gel and molecular weight standard:
- Agarose gel: 4% E-Gel® (Cat. No. G5000-04)
- Molecular weight standard: 10 bp DNA Ladder (Cat. No. 10821-015)
When analyzing an aliquot of the annealed ds oligo reaction by agarose gel electrophoresis, we generally see the following:
- A detectable higher molecular weight band representing annealed ds oligo.
- A detectable lower molecular weight band representing unannealed single-stranded oligos. Note that this band is detected since a significant amount of the single-stranded oligo remains unannealed.
Please see the image below for an example of expected results:

For optimal results, use a 10:1 molar ratio of ds oligo insert:vector for ligation.
Please follow the steps outlined below:
- Visit RNAi Designer
- Enter an accession number or provide a nucleotide sequence
- Determine the region for target design: ORF, 5’ UTR, or 3’ UTR
- Choose database for Blast
- Choose minimum and maximum G/C percentage
Select vector and strand orientation and click “RNAi Design” to design shRNA
Transcription of the shRNA initiates at the first base following the end of the U6 promoter sequence. In the top-strand oligo, the transcription initiation site corresponds to the first nucleotide following the 4 bp CACC sequence added to permit directional cloning. We recommend initiating the shRNA sequence at a guanosine (G) because transcription of the native U6 snRNA initiates at a G. Note the following:
- If G is part of the target sequence, then incorporate the G into the stem sequence in the top-strand oligo and add a complementary C to the 3′ end of the top-strand oligo.
- If G is not the first base of the target sequence, we recommend adding a G to the 5′ end of the top-strand oligo directly following the CACC overhang sequence. In this case, do not add the complementary C to the 3′ end of the top-strand oligo. Note: We have found that adding the complementary C in this situation can result in reduced activity of the shRNA. Alternative, if use of a G to initiate transcription is not desired, use an adenosine (A) rather than C or T. Note, however, that use of any nucleotide other than G may affect initiation efficiency and position.
You can use a loop sequence of any length ranging from 4 to 11 nucleotides, although short loops (i.e., 4–7 nucleotides) are generally preferred. Avoid using a loop sequence containing thymidines (Ts), as they may cause early termination. This is particularly true if the target sequence itself ends in one or more T nucleotides. Here are some loop sequences we recommend:
- 5’ – CGAA – 3’
- 5’ – AACG – 3’
- 5’ – GAGA – 3’
Unfortunately, the pENTR/U6 vector does not contain a selection marker; therefore, only transient RNAi analysis may be performed. If you wish to generate stable cell lines, perform an LR reaction into an appropriate Gateway® destination vector to generate expression clones.
The pENTR™/H1/TO vector contains the Zeocin™ resistance gene to facilitate generation of cell lines that inducbily express the shRNA of interest. Perform a kill curve to determine the minimum concentration of Zeocin™ that is required to kill your untransfected mammalian cell line. Please note that Zeocin™-sensitive cells do not round up and detach from the plate, but rather may increase in size, show abnormal cell shape, display presence of large empty vesicles in the cytoplasm, or show breakdown of plasma/nuclear membranes.
Yes, our pENTR™/H1/TO construct is a tetracycline-regulated shRNA expression, based on a repression/derepression mechanism. The amount of Tet repressor (TR) expressed in the host cell line will determine the level of transcriptional repression of the Tet operator sequences in your pENTR™/H1/TO construct. The mechanism of tetracycline regulation in the system is based on the binding of tetracycline to the Tet repressor and derepression of the promoter controlling expression of the shRNA of interest. In the system, expression of your shRNA of interest is repressed in the absence of tetracycline and induced in its presence. In the BLOCK-iT™ Inducible H1 RNAi Entry Vector System, expression of the shRNA of interest from the pENTR™/H1/TO is controlled by a human H1 promoter into which 2 copies of the 19-nucleotide Tet operator sequence (TetO2) have been incorporated (i.e., H1/TO promoter). Each 19-nucleotide TetO2 sequence serves as the binding site for 2 molecules of the Tet repressor.
In the absence of tetracycline, the Tet repressor (expressed from the pcDNA™6/TR plasmid or pLenti6/TR lentiviral construct, as desired) forms a homodimer that binds with extremely high affinity to each TetO2 sequence (Hillen & Berens, 1994) in the H1/TO promoter of the pENTR™/H1/TO. The two TetO2 sites in the H1/TO promoter serve as binding sites for 4 molecules (or 2 homodimers) of the Tet repressor (see figure on the next page). Binding of the Tet repressor homodimers to the Tet O2 sequences represses transcription of your shRNA of interest. Upon addition, tetracycline binds with high affinity to each Tet repressor homodimer in a 1:1 stoichiometry and causes a conformational change in the repressor that renders it unable to bind the Tet operator. The Tet repressor:tetracycline complex then dissociates from the Tet operator and allows induction of transcription of the shRNA of interest, resulting in target gene knockdown.
Note: The affinity of the Tet repressor for the Tet operator is KB = 2 x 1011 M–1 (as measured under physiological conditions), where KB is the binding constant (Hillen & Berens, 1994). The association constant, KA, of tetracycline for the Tet repressor is 3 x 109 M-1 (Takahashi et al., 1991).
We do not offer an anti-TR antibody. Even though a western blot using an anti-TR antibody can be used to screen out clones that do not express any TR protein, it would not be the optimal way to screen for functional clones. Functional testing by doing a transient transfection with the lacZ expression control plasmid is recommended for this purpose, followed by picking a clone that shows the lowest basal levels of expression of β-galactosidase in the absence of tetracycline and highest levels of β-galactosidase expression upon addition of tetracycline.
Doxycycline may be used as an alternative inducing agent in the BLOCK-iT™ Inducible H1 RNAi System. It is similar to tetracycline in its mechanism of action, and exhibits similar dose response and induction characteristics as tetracycline in the T-REx™ System. Doxycycline has been shown to have a longer half-life than tetracycline (48 hours vs. 24 hours, respectively). We do not offer doxycycline, but it may be obtained from Sigma (Cat. No. D9891).
A dose response curve or kill curve is a simple method for determining the optimal antibiotic concentration to use when establishing a stable cell line. Untransfected cells are grown in a medium containing antibiotic at varying concentrations in order to determine the lowest amount of antibiotic needed to achieve complete cell death. The basic steps for performing a dose response curve or kill curve are as follows:
- Plate untransfected cells at 25% confluence, and grow them in a medium containing increasing concentrations of the antibiotic. For some antibiotics, you will need to calculate the amount of active drug to control for lot variation.
- Replenish the selective medium every 3–4 days. After 10–12 days, examine the dishes for viable cells. The cells may divide once or twice in the selective medium before cell death begins to occur.
- Look for the minimum concentration of antibiotic that resulted in complete cell death. This is the optimal antibiotic concentration to use for stable selection.
Please follow the directions listed on page 52 of the manual.
No, you should use an entry vector that contains the elements necessary for RNA Polymerase III–dependent expression of your shRNA (i.e., Pol III promoter and terminator).
Please visit our Transfection Support Center to find technical resources, tips and tricks, and troubleshooting information on transfection.
miRNA RNAi Vectors
MicroRNAs (miRNAs) are endogenously expressed small single stranded RNA sequences of ~22 nucleotides in length which naturally direct gene silencing through components shared with the RNAi pathway (Bartel, 2004). Unlike shRNAs, however, the miRNAs are found embedded, sometimes in clusters, in long primary transcripts (pri-miRNAs) of several kilobases in length containing a hairpin structure and driven by RNA Polymerase II (Lee et al., 2004), the polymerase also responsible for mRNA expression. Drosha, a nuclear RNase III, cleaves the stem-loop structure of the pri-miRNA to generate small hairpin precursor miRNAs (pre-miRNAs) which are ~70 nucleotides in length (Zeng et al., 2005). The pre-miRNAs are exported from the nucleus to the cytoplasm by exportin-5, a nuclear transport receptor (Bohnsack et al., 2004; Yi et al., 2003). Following the nuclear export, the pre-miRNAs are processed by Dicer into a ~22 nucleotides miRNA (mature miRNA) molecule, and incorporated into an miRNA-containing RNA-induced silencing complex (miRISC) (Cullen, 2004).
Please see the comparison chart below of our RNAi vector technologies:
| BLOCK-iT™ Pol II miR RNAi | BLOCK-iT™ shRNA |
Pol II expression system | Pol III expression system |
Polycistronic expression of miRNAs (chaining) | Single shRNA expression per promoter |
Co-expression of GFP allows expression to be tracked | Only delivery can be tracked (with separate expression cassette) |
Compatible with most Gateway® DEST vectors | Compatible with BLOCK-iT™ DEST vectors |
Best design success (>70%) | Moderate design success (~50%) |
These vectors can be used for stable expression and the ability to use viral delivery. These miR RNAi vectors include flanking and loop sequences from an endogenous miRNA which directs the excision of the engineered miRNA from a longer Pol II transcript (pri-miRNA). When present in the nucleus, these vectors efficiently use the endogenous cellular machinery to process knockdown sequences that are specifically designed to have 100% homology to your target of interest and will result in target cleavage. In addition, the loop sequence has a unique restriction site, so that it can be linearized for more efficient sequencing, sometimes a challenge with standard shRNA hairpins. The kits offer over 70% knockdown success, easy expression tracking (with co-cistronic expression of Green Fluorescent Protein), multiple target knockdown, and constitutive or inducible expression.
The vectors are designed and optimized for expressing modified miR155 structure, and finally make siRNA for gene targeting using RNAi pathways. Additional optimization might be necessary to express native miRNA. Alternatively, the sequences of native miRNA can be cloned into a standard protein expression vector and inspect for miRNA production. Please see our two suggestions for miRNA overexpression:
1. Use PCR on gDNA to amplify the endogenous pre-miRNA hairpin as well as ~50–80 bp of flanking sequences on each side, then TOPO clone into an expression vector such as one of our pcDNA vectors. This will produce a transcript which contains the pre-miRNA in the context of its natural flanking sequences. This will probably be the best mimic of the endogenous miRNA, because the flanking sequences and precursor have the information needed to correctly process out the mature miRNA. The disadvantage of this technique is that it is somewhat laborious and wouldn’t be as amenable to looking at many miRNAs (each one requiring identification of the genomic locus, primer design, and successful PCR).
2. Use the mature miRNA sequence (or the first 21 nucleotides of it) as the “antisense” sequence in Invitrogen’s BLOCK-iT™ Pol II miR RNAi vector system. This technique has been successfully published (see Lee et al., PNAS 2006;103;15669-15674) and is quick and simple for design and cloning.
Using this method, many miRNA vectors could be built the same way. We also have a fairly good understanding that the major product of the miR RNAi vectors has the expected 5’ end for the mature miRNA. However, we also know that the 3’ end is variable and includes a number of slightly smaller and longer species that can include nucleotides from the loop (GUU…). The 3’ end is probably least critical to miRNA function, but there may be some miR:target interactions for which it is important, and we just don’t know how closely these mimic endogenous miRNAs.
We would recommend use of our pcDNA6.2™GW/EmGFP-miR vector, where EmGFP is expressed co-cistronically with your miRNA of interest. You should see 100% correlation of EmGFP expression with the knockdown activity of your miRNA. Please see the example below:

In this example, cells were specifically transfected with Lipofectamine® 2000 reagent at an expected 50% efficiency to demonstrate the 100% tracking of EmGFP and miRNA expression. After 48 hours, cells were stained with Hoechst nuclear stain (which stains all cells), stained with a red lamin stain, and monitored for GFP expression. Nearly half of the cells highly express the lamin protein. When cells expressing EmGFP and lamin A/C stained are combined, it is clear that cells expressing GFP do not appear to have lamin A/C present, and cells stained red for lamin A/C do not appear to have any GFP expression. This demonstrates that cells expressing EmGFP are also all greatly reduced in lamin expression due to the presence of the miRNA that is co-cistronically expressed.
The EmGFP from the pcDNA™6.2-GW/EmGFP-miR expression vector has the following excitation and emission wavelengths, as published in the literature (Tsien, 1998): 487 nm and 509 nm, respectively. Detection can be performed using a standard FITC filter set. We recommend Omega XF100.
miRNAs are sometimes expressed in clusters in long primary transcripts driven by RNA Pol II (Lee et al., 2004). Our vectors support chaining of miRNAs to express them in one primary transcript, thus ensuring co-cistronic expression of multiple miRNAs. In the final construct, the original pattern of restriction sites is regenerated, making the construct amenable to multiples rounds of chaining. The figure below shows the principle of chaining two miRNAs, derived from two different miRNA vectors, into one miRNA expression vector. Note: Chaining together miRNAs targeting different genes usually results in slightly reduced knockdown of each gene. Chaining different miRNAs targeting the same gene or repeating one miRNA can enhance knockdown. Due to increased processing, EmGFP expression is attenuated by miRNA chaining.

See page 33 of the manual for directions on how to chain pre-miRNAs.
We would expect this G to be critical, as it is part of the last base pair in the “flanking” region (derived from mouse miR-155) before the start of the mature miRNA. The mature miRNA sequence will act as the guide strand for the RNAi.
Please see the table below for Gateway® destination vector compatibility:
| Destination Vector or System | Compatibility | Cat. No. |
ViraPower™ Lentiviral Vectors, including Multisite Gateway® | Compatible with pLenti6/V5-DEST™, pLenti6/UbC/V5-DEST™ | V49610, V49910, K493400 |
EF-1a promoter (pEF-DEST51) | Yes | 12285011 |
T-REx™ (pT-REx™-DEST30) | Yes | 12301016 |
Flp-In™ (pEF5/FRT/V5-DEST™) | Yes | V602020 |
N-terminal reporter tags (GeneBLAzer™, YFP, Lumio™) | Compatible with pcDNA™6.2/N-YFP/DEST | V358-20 |
MultiSite Gateway® (pDEST/R4-R3) | Compatible with TK polyA 3’ element and various 5’ promoter elements | 12537023 |
Please note, transferring the pre-miRNA expression cassette from pcDNA™6.2-GW/EmGFP-miR to the pLenti6/BLOCK-iT™-DEST vector will not yield in a functional miRNA expression vector. Expression of the pre-miRNA requires the destination vector to supply a Pol II promoter.
Please visit our BLOCK-iT® RNAi Designer and select miR RNAi as your target design option. This miR RNAi can then be cloned into the pcDNA™6.2-GW/miR and pcDNA™6.2/EmGFP-miR vectors.
In order to create a scramble miR RNAi negative control we recommend keeping 2-3 nt on each end of the guide RNA the same and scrambling the middle, then conducting BLAST to look for obvious problems. In order to create a point mutation miR RNA negative control, a single change may not be enough, but the best place to put it would be at nt 10 or 11 of the antisense sequence.
The pcDNA™6.2-GW/+EmGFP-miR expression construct contains the Basticidin resistance gene to allow for Blasticidin selection of mammalian cells that are stably transfected with the pcDNA™6.2-GW/+EmGFP-miR construct. Start by performing a kill curve on your untransfected mammalian cells, followed by transfection of your expression clone into the mammalian cell line of choice and selecting for stable cell lines using Blasticidin.
Perform a DraI digestion and self ligation of the vector to form a pcDNA™6.2-GW/miR clone expressing the same pre-miRNA. See page 40 of the manual for a more detailed protocol.
Virus-Based RNAi Expression Systems
Adenoviruses are DNA viruses that can transiently transduce nearly any mammalian cell type. The adenovirus enters target cells by binding to the coxsackie adenovirus receptor (CAR). After binding to the CAR, the adenovirus is internalized via integrin-mediated endocytosis followed by active transport to the nucleus, where its DNA is expressed episomally.
Oncoretroviruses and lentiviruses are positive-strand RNA viruses that stably integrate their genomes into host cell chromosomes. When pseudotyped with an envelope that has a broad tropism, such as vesicular stomatitis virus glycoprotein (VSV-G), these viruses can enter virtually any mammalian cell type. However, the oncoretroviruses depend upon nuclear membrane breakdown during cell division to transduce cells. In contrast, lentiviruses are more versatile tools, as they use an active nuclear import pathway to transduce nondividing cells.
Adenoviral Systems | Lentiviral Systems |
Efficiency transduce both dividing and nondividing cells | Efficiently transducer both dividing and nondividing cells |
Study gene knockdown with high-level transient expression | Study long-term gene knockdown with stable expression |
Reliably and reproducibly transducer cell populations | Reproducibly transducer cell populations |
Inducible or constitutive gene knockdown | Inducible or constitute gene knockdown |
It should be noted that gene expression from both systems is typically detected within 24–48 hours of transduction, so both systems can be used for experiments of a transient nature. The main difference is that lentivirus integrates into the host genome and adenovirus does not. Higher viral titers are achieved with the adenovirus. Lentiviral delivery systems are available for both shRNA and miR RNAi vectors, and an adenoviral delivery system is available for shRNA vectors.
Please see the steps below:
- Clone the double-stranded DNA oligo encoding an shRNA or miR RNAi into one of the BLOCK-iT™ entry (shRNA) or expression (miR RNAi) vectors.
- Transfer the RNAi cassette into the adenoviral (shRNA only) or lentiviral destination vector by Gateway® recombination.
- Transfect RNAi vectors into the viral producer cells to produce viral stocks, which can be used immediately or stored at –80°C.
- Harvest viral supernatants and determine the titer (amplify adenoviral stocks if desired).
- Transduce lentiviral or adenoviral stocks to any cell type.
Please see the definitions below:
- Infection: Applies to situations where viral replication occurs and infectious viral progeny are generated. Only cell lines that stably express E1 can be infected.
- Transduction: Applies to situations where no viral replication occurs and no infectious viral progeny are generated. Mammalian cell lines that do not express E1 are transduced. In this case, you are using adenovirus as a vehicle to deliver shRNA.
Adenovirus is not an actively lytic virus, meaning that mature viral particles accumulate in the cell over the course of two to three days. As virus accumulates, the producer cell rounds up and eventually bursts due to the sheer number of virus particles inside. Once this occurs, neighboring cells become infected and the three-day cycle begins again. The term “cytopathic effect”, or CPE, is used to describe this and is typically visible within ~7 days posttransfection in the form of “comet-shaped” plaques resulting from two rounds of infection, replication and cell burst.
- After 7 days, CPE will expand and eventually take over the plate by ~10 days posttransfection.
- ~10 days are required to produce virus from a transfected dish of cells (as just described).
- Once an initial viral stock is produced, it can be amplified directly by infection of fresh 293A cells at a multiplicity of infection (MOI) of 3.
It is strongly recommended that the lentivirus or adenovirus stocks be aliquoted immediately after production and stored at _80°C. Lentivirus is more sensitive to freeze/thaw than adenovirus. Adenovirus stocks can typically be frozen/thawed up to 3 times without loss of titer, while lentivirus stocks can lose up to 5% or more of their titer with each freeze/thaw. When stored properly, viral stocks can be used for up to one year. After long-term storage, please retiter your viral stocks before use.
BLOCK-iT™ RNAi Lentiviral Systems
Lentivirus is a genus of slow retroviruses, characterized by a long incubation period.
Our lentiviral vectors are based on the HIV-1 backbone. However, several alterations have been made so they function solely as a gene delivery vehicle without subsequent viral replication or disease. Specific HIV-1 genes have been deleted to enhance safety. The HIV-1 genes are only expressed in the producer cells (293FT) and none of them are packaged into the viral genome and thus are never expressed in the transduced target cell.
We recommend storing lentiviral expression vectors at –20°C. Due to their relatively large size, we do not recommend storing these vectors at –80°C, as the vector solution will completely freeze, and too many freeze/thaws from –80°C will affect the cloning efficiency.
Use of the BLOCK-iT™ Lentiviral RNAi Expression System to facilitate lentiviral based delivery of shRNA to mammalian cells provides the following advantages:
- The pENTR™/U6 entry vector provides a rapid and efficient way to clone ds oligo duplexes encoding a desired shRNA target sequence into a vector containing an RNA Pol III-dependent expression cassette (i.e., U6 RNAi cassette) for use in RNAi analysis.
- The vectors in the System are Gateway®-adapted for easy recombination of the U6 RNAi cassette from the pENTR™/U6 vector into the pLenti6/BLOCK-iT™-DEST vector.
- Generates a replication-incompetent lentivirus that effectively transduces both dividing and nondividing mammalian cells, thus broadening the potential RNAi applications beyond those of other traditional retroviral systems (Naldini, 1998).
- Efficiently delivers the shRNA of interest to mammalian cells in culture or in vivo.
- Provides stable, long-term expression of the shRNA of interest beyond that offered by traditional adenovirus-based systems.
- Produces a pseudotyped virus with a broadened host range (Yee, 1999).
- Includes multiple features designed to enhance the biosafety of the system.
We do not recommend using miniprep plasmid DNA for lentivirus production. We recommend preparing lentiviral plasmid DNA using the S.N.A.P.™ MidiPrep Kit (Cat. No. K1910-01) or PureLink® HiPure Plasmid Midiprep Kit (Cat. No. K210004), which contain 10 mM EDTA in the Resuspension Buffer. Since lentiviral DNA midipreps also often have low DNA yields, we recommend following specific protocols to increase yield—basically, grow cells slowly, use fewer cells per column, and use 100 mL lentiviral culture for each DNA midiprep.
Yes, the lentiviral expression vector will work as an expression vector by itself and can be stably selected with the appropriate antibiotic. Please note that the vector will be about twice the size of most regular vectors. Therefore you may need to increase the amount of transfected vector to approximate molar equivalents.
Constitutive knockdown is virtually identical for these two promoters in HEK 293 cells. In other cell types there are reports that either H1 or U6 may be more active, though in general, differences are minimal.
The packaging mix contains an optimized mixture of the three packaging plasmids: pLP1, pLP2, and pLP/VSVG. These plasmids supply the helper functions as well as structural and replication proteins in trans required to produce the lentivirus. Unfortunately, we do not sell the packaging plasmids separately; however, you can purchase the ViraPower™ Packaging Mix as a stand-alone product (Cat. No. K497500).
The lentiviral and packaging vectors supplied in the BLOCK-iT™ Lentiviral RNAi Expression System are third-generation vectors based on lentiviral vectors developed by Dull et al., 1998. This third-generation lentiviral system includes a significant number of safety features designed to enhance its biosafety and to minimize its relation to the wild-type human HIV-1 virus. The BLOCK-iT™ Lentiviral RNAi Expression System includes the following key safety features:
- The pLenti6/BLOCK-iT™-DEST expression vector contains a deletion in the 3’ LTR (ΔU3) that does not affect generation of the viral genome in the producer cell line, but results in “self-inactivation” of the lentivirus after transduction of the target cell (Yee et al., 1987; Yu et al., 1986; Zufferey et al., 1998). Once integrated into the transduced target cell, the lentiviral genome is no longer capable of producing packageable viral genome.
- The number of genes from HIV-1 that are used in the system has been reduced to three (i.e., gag, pol, and rev).
- The VSV-G gene from Vesicular Stomatitis Virus is used in place of the HIV-1 envelope (Burns et al., 1993; Emi et al., 1991; Yee et al., 1994).
- Genes encoding the structural and viral genome packaging components are separated onto four plasmids. All four plasmids have been engineered not to contain any regions of homology with each other to prevent undesirable recombination events that could lead to the generation of a replication competent virus (Dull et al., 1998).
- Although the three packaging plasmids allow expression in trans of proteins required to produce viral progeny (e.g., gal, pol, rev, env) in the 293FT producer cell line, none of them contain LTRs or the Ψ packaging sequence. This means that none of the HIV-1 structural genes are actually present in the packaged viral genome, and thus, are never expressed in the transduced target cell. No new replication-competent virus can be produced.
- The lentiviral particles produced in this system are replication-incompetent and only carry the gene of interest. No other viral species are produced.
- Expression of the gag and pol genes from pLP1 has been rendered Rev dependent by virtue of the HIV-1 RRE in the gag/pol mRNA transcript. Addition of the RRE prevents gag and pol expression in the absence of Rev (Dull et al., 1998).
- A constitutive promoter (RSV promoter) has been placed upstream of the 5’ LTR in the pLenti6/BLOCK-iT™-DEST expression vector to offset the requirement for Tat in the efficient production of viral RNA (Dull et al., 1998).
Despite the inclusion of the safety features discussed on the previous page, the lentivirus produced with this system can still pose some biohazard risks since it can transduce primary human cells. For this reason, we highly recommend that you treat lentiviral stocks generated using this system as Biosafety Level 2 (BL-2) organisms and strictly follow all published BL-2 guidelines with proper waste decontamination. Furthermore, exercise extra caution when creating lentiviruses carrying potential harmful or toxic genes (e.g., activated oncogenes). For more information about the BL-2 guidelines and lentivirus handling, refer to the document “Biosafety in Microbiological and Biomedical Laboratories,”
5th Edition, published by the Centers for Disease Control and Prevention (CDC). This document may be downloaded here.
Our lentiviral expression vectors contain approximately 20% of the original viral genome. The rest of the viral genome is deleted from our lentiviral expression vectors for safety reasons.
Our lentiviral expression vectors belong to the third-generation, meaning that we use a four plasmid vector system (1 lentiviral expression vector and 3 packaging plasmids), thus eliminating concerns about recombination events bringing components together as a single vector to produce replication-competent lentivirus. Further, these vectors contain a chimeric 5’ LTR, by means of which, virus production is not dependent on the HIV tat transactivator. Also, the original U3 region of the LTR (Long Terminal Repeat) is deleted to make the virus self-inactivating and thus replication-incompetent.
Our lentiviral packaging mix belongs to the third generation, meaning that it does not express the tat gene. Further, gag/pol, and rev genes are supplied as independent plasmids, thus eliminating concerns about recombination events bringing components together as a single vector to produce replication-competent lentivirus.
Our lentiviral expression vectors belong to the third generation, meaning that they contain a chimeric 5’ LTR, by means of which, virus production is not dependent on the HIV tat trans-activator. As a result, they are compatible with a second- or third-generation packaging mix.
Your second-generation lentiviral vector will not be compatible with our packaging mix because our packaging mix belongs to the third generation, meaning that it does not express the tat gene whereas your lentiviral vector will need the tat gene for virus production.
Our lentiviral packaging mix belongs to the third generation, meaning that it does not express the tat gene. It can be used with lentiviral vectors that belong to the third generation or higher, where virus production is independent of the tat gene.
For optimal results, use Stbl3™ E. coli for transformation, as this strain is particularly well-suited for use in cloning unstable DNA such as lentiviral DNA, which contains direct repeats. You may transform the LR recombination reaction into other recA, endAE. coli strains, including TOP10 and DH5α™, if desired. Note, however, that these strains are not as well-suited for cloning unstable DNA, and may give rise to a low percentage (< 5%) of transformants containing unwanted recombinants (i.e., plasmids where recombination has occurred between the 5’ and 3’ LTRs) when selected on plates containing only ampicillin. These events occur less frequently when selection is performed using both ampicillin (100 μg/mL) and blasticidin (50 μg/mL). Note also that transformed E. coli grow more slowly in LB media containing ampicillin and blasticidin, and may require slightly longer incubation times to obtain visible colonies.
The “F” stands for the high transfection efficiency of this particular 293 cell clone (called 293F), and the “T” stands for the SV40 large T antigen. The large T antigen expression plasmid is stably integrated in the genome and confers resistance to Geneticin® antibiotic in these cells. The presence of the SV40 large T antigen is important for high-titer lentivirus production and the mechanism is not known. If regular 293 cells or another 293T cell line is used as the producer cell line, you will be able to produce virus, but the titers will be lower. Please note that since 293FT cells stably express the SV340 large T antigen, we do not recommend generating stable cell lines with plasmids that contain the SV40 origin of replication.
We use mycoplasma-tested Gibco® FBS (Cat. No. 16000-044) and use the following plasticware for 293FT cells: We have observed that when 293FT cells are cultured in the presence of this FBS following the instructions in the manual, virus production is better than that obtained with many other serum sources.
- T175--Fisher Cat. No. 10-126-13; this is a Falcon flask with 0.2 μm vented plug seal cap.
- T75--Fisher Cat. No. 07-200-68; this is a Costar flask with 0.2 μm vented seal cap.
- 100 mm plate--Fisher Cat. No. 08-772E; this is a Falcon tissue culture-treated polystyrene plate.
We get excellent adherence on these plates under routine cell culture/maintenance conditions.
The 293FT cell line stably expresses the SV40 large T antigen from the pCMVSPORT6Tag.neo plasmid that contains the neomycin resistance marker. In order to maintain the plasmid/phenotype, the cells have to be routinely cultured in medium containing Geneticin® (G418) antibiotic at a concentration of 500 μg/mL.
During virus production, the following observations should be made:
- 293FT change their morphology
- They are multi-nucleated (syncytia)
- They start to look like balloons or as if they are about to explode
- They often, but not always, lift off from the surface
- Untransfected 293FT cells leave empty spaces and “pile” up at other spots of the flask.
View the pictures below of 293FT cells pre-transfection, 6 hr post-transfection, 24 hr post-transfection, and 48 hr post-transfection.




We recommend aliquoting lentiviral stocks immediately after production, into small working volumes and storing at –80°C for long-term storage. Lentivirus is sensitive to storage temperature and to freeze/thaw and should be handled with care. It can lose up to 5% or more activity with each freeze/thaw. When stored properly, viral stocks of an appropriate titer should be suitable for use for up to 1 year. After long-term storage, we recommend retitering your viral stocks before use.
Lentivirus produced using our system is replication-incompetent, and this is a safety feature. You must perform a fresh transfection each time you need more virus.
Lentiviral Expression System- Viral Titering
We highly recommend performing a viral titer as it helps to:
- Establish optimal transduction rate for a given cell
- Control the number of integrated copies of the lentivirus
- Generate reproducible expression results
We strongly recommend the human fibrosarcoma cell line HT1080 (ATCC, Cat. No. CCL-121) as the “gold standard” for titering lentivirus. The primary reason is that transduction efficiency is high in these cells, and titering results are very accurate and reproducible. However, you may also use the same mammalian cell line to titer your lentiviral stocks as you will use to perform your expression studies. In general, this should be an adherent, nonmigratory cell line, and exhibit a doubling time in the range of 18–25 hours. Regular 293 cells may be used for lentivirus titering but we do not recommend using 293T or 293FT cells because these cells contain the SV40 large T antigen that will induce unwanted DNA replication at the SV40 ori contained within the integrated lentiviral expression vector. This often leads to cell death and results in very low titers.
- For the 293FT cells, use a low passage (<20). Passage cells in complete DMEM containing G418 (500 µg/mL). Supplement the medium with “nonessential” amino acids and sodium pyruvate. Plate cells at 5 x 106 per 100 mm dish. When plating for transfection the next day, do not add any antibiotics to the medium.
- When working with Lipofectamine® 2000 reagent and your plasmid DNA, make sure the mix is treated gently—do not pipet up and down. Ensure that plasmid DNA used has been isolated with Invitrogen™ S.N.A.P. columns (midiprep) or PureLink® maxi or mega kits and is sufficiently clean for these transfections. Do not use plasmid DNA from minipreps.
You can use the p24 ELISA assay for lentivirus titering, but keep in mind that the titer will not be a measure of functional virus since it will measure both inactive and active virus. As a result, titers obtained using this method are usually about 10-fold higher than with methods that measure functional virus such as blasticidin selection.
You can perform PCR/qPCR analysis of viral genes for lentivirus titering, but keep in mind that the titer will not be a measure of functional virus since it will measure both inactive and active virus. As a result, titers obtained using this method are usually about 10-fold higher than with methods that measure functional virus such as blasticidin selection.
Yes, FACS analysis can be performed for GFP expression to determine the titer. If the lenti vector does not contain GFP, use the antibiotic selection titering protocol. With lacZ-expressing lentivirus, one can get an approximate idea of the titer (based on lacZ expression following transductions at different viral dilutions), but this is not a very quantitative titering method. Based on our experience we have found that LacZ titering is not nearly as accurate or reproducible as titering based on GFP expression or Bsd selection, since one has to rely on counting blue cells by eye under the microscope.
Lentiviral Expression System—Transduction and Analysis
The lentiviral envelope is pseudotyped with the vesicular stomatitis virus glycoprotein (VSV-G), which allows the lentivirus to interact with its target cell in a receptor-independent manner. As a result, lentivirus has broad tropism and can, in theory, transduce any mammalian cell type.
This receptor-independent entry into the target cell likely involves endocytosis (Espenshade et al. (2002) Proc Natl Acad Sci USA 99:11694; Aiken (1997) J Virol 71:5871.)
We have found that, in general, 80–90% of the cells in an actively dividing cell line (e.g., HT1080) express a target gene when transduced with lentivirus at an MOI of ~1. Some nondividing cell types transduce lentiviral constructs less efficiently. For example, only about 50% of the cells in a culture of primary human fibroblasts express a target gene when transduced at an MOI of ~1. If you are transducing your lentiviral construct into a nondividing cell type, you may need to increase the MOI (e.g., MOI = 10) to achieve optimal expression levels for your recombinant protein. If you are transducing your lentiviral construct into your mammalian cell for the first time, we recommend using a range of MOIs (e.g.,0, 0.5, 1, 2, 5, 10) to determine the MOI required to obtain the optimal protein expression for your application.
MOI stands for multiplicity of infection. MOI is defined as the number of virus particles per cell and generally correlates with the number of integration events and, as a result, expression. Typically, expression levels increase as the MOI increases.
A number of factors influence optimal MOI which include:
- The nature of your mammalian cell line (i.e., nondividing vs. dividing cells)
- The transduction efficiency of your mammalian cell line
- The nature of your target gene of interest
- The procedure used to express your shRNA
As a starting point, transduce the Lenti6/TR construct into cells at an MOI of 10, and transduce the
Lenti4/BLOCK-iT™-DEST construct into cells at an MOI of 1 to 5. You may optimize basal and tetracycline-induced expression levels by varying the MOI of the Lenti6/TR and/or Lenti4/BLOCK-iT™-DEST lentiviruses transduced.
Polybrene® reagent (hexadimethrine bromide) is a cationic polymer available from Sigma-Aldrich (Cat. No. H9268) that increases transduction efficiency by neutralizing the charge repulsion between virus particles and the cell surface. For best results, we recommend performing transduction in the presence of Polybrene® reagent. Note, however, that some cells are sensitive to Polybrene® reagent (e.g.,primary neurons). Hence, before performing a transduction experiment, you may want to test your cell line for sensitivity to Polybrene® reagent at a range of 0–10 μg/mL, and avoid using Polybrene® reagent if the cells exhibit toxicity or phenotypic changes.
Please try using DEAE-dextran. See the following reference, in which cells were transduced with DEAE:
Reiser et al. (1996) Proc Natl Acad Sci USA, 93:15271.
Yes, it is possible to concentrate VSV-G pseudotyped lentiviruses using a variety of methods without significantly affecting their transducibility. If the titer of your lentiviral stock is relatively low (less than 5 x 105 TU/mL) and your experiment requires that you use a large volume of viral supernatant (e.g., a relatively high MOI), you may wish to concentrate your virus before proceeding to transduction. Please refer to the reference Yee, J.K., The Development of Human Gene Therapy 1999, pp. 21–45, for guidelines on how to concentrate your virus.
Integration of the lentivirus into the genome is random. Depending upon the influence of the surrounding genomic sequences at the integration site, you may see varying levels of target gene knockdown from different blasticidin- and Zeocin™-resistant clones. Test at least 10 blasticidin- and Zeocin™-resistant clones, and select the clone that provides the lowest basal and the highest level of induced target gene knockdown for further studies.
When performing RNAi studies using pLenti4/BLOCK-iT™-DEST lentiviral constructs, we generally observe significant inhibition of gene expression within 48–120 hours after transduction. The degree of gene knockdown depends on the time of assay, stability of the protein of interest, and on the other factors. Note that 100% gene knockdown is generally not observed, but >80% is possible with optimized conditions.
Yes, we offer 4 T-REx™ cell lines: 293, HeLa, CHO, or Jurkat cells. Alternatively, you can create your own T-REx™ cell line that stably expresses the Tet repressor form the pcDNA™6/TR.
Stbl3 Competent Cells
Stbl3 cells were derived from the HB101 strain.
The specific genetic marker that allow for reduced recombination has not been identified.
Stbl3 is specifically recommended and tested for cloning of unstable lentiviral DNA sequences containing direct repeats, including all Invitrogen™ pLenti expression vectors. They have not been tested for stabilization of other sequences, but may be effective in some cases where Stbl2 or Stbl4 cells have not worked.
A periplasmic endonuclease (Endonuclease A) can co-purify with the plasmid DNA under certain conditions, which could result in degradation of the isolated DNA in downstream applications. Endonuclease A is very stable, and boiling will not guarantee removal. Often, the presence of endonuclease does not become clear until the plasmid DNA is incubated at warmer temperatures, such as in a restriction digest. The endonuclease becomes very active at 37°C, and when the digested plasmid is later analyzed on a gel, only a smear of degraded DNA appears.
The following procedures are recommended to ensure removal of endonuclease from Stbl3 plasmid preps:
- If using a silica-based column for purification, look for the presence of an “Optional Wash” step in the procedure. (Example: Wash Buffer W10 in the PureLink® Quick Plasmid Miniprep Kit, Cat. No. K2100-10). This “optional” wash is absolutely necessary for purifying plasmids from Stbl3 cells.
- For larger-scale plasmid preps using an anion-exchange column (such as PureLink® HiPure), endonuclease is generally removed from the DNA during the normal procedure.
- If making solutions I, II, III for a conventional miniprep procedure, make sure EDTA is present in solution I. The endonuclease needs magnesium for activity, and EDTA will remove Mg++. Resuspend the plasmid DNA in TE buffer.
- In any of these methods, an additional phenol/chloroform extraction of purified DNA would be recommended to be sure that endonuclease has been removed from the purified plasmid.
Adenovirus Expression System
The vector contains the elements required to allow packaging of the expression construct into virions (e.g., 5’ and 3’ ITRs, encapsidation signal, adenoviral late gene). Specifically, the pAd/BLOCK-iT™-DEST vector contains:
- Human adenovirus type 5 sequences (Ad 1-458 and 3513-35935) encoding genes and elements (e.g., Left and Right Inverted Terminal Repeats (ITRs), encapsidation signal sequence, late genes) required for proper packaging and production of adenovirus (Hitt et al., 1999; Russell, 2000)
- Two recombination sites, attR1 and attR2, for recombinational cloning of the U6 RNAi cassette from the pENTR™/U6 entry clone using Gateway® technology
- Chloramphenicol resistance gene (CmR) located between the two attR sites for counter selection
- The ccdB gene located between the attR sites for negative selection
- Ampicillin resistance gene for selection in E. coli
- pUC origin for high-copy replication and maintenance of the plasmid in E. coli
Adenovirus enters target cells by binding to the coxsackie/adenovirus receptor (CAR) (Bergelson et al., 1997). After binding to the CAR, the adenovirus is internalized via integrin-mediated endocytosis (Russell, 2000) followed by active transport to the nucleus. Once in the nucleus, the early events are initiated (e.g., transcription and translation of E1 proteins), followed by expression of the adenoviral late genes and viral replication. Note that expression of the late genes is dependent upon E1. In the BLOCK-iT™ Adenoviral RNAi Expression System, the 293A producer cells supply E1. The viral life cycle spans approximately 3 days. For more information about the adenovirus life cycle and adenovirus biology, refer to published reviews (Russell, 2000).
Human Adenovirus Type 5 (Ad5) enters target cells via the coxsackie virus and adenovirus receptor (CAR), followed by an integrin-mediated internalization mechanism. CAR/integrin proteins are ubiquitously present on mammalian cells, thus affording adenovirus the ability to transduce a very broad range of cell types. If your specific cell type has very low expression of CAR, adenoviral transduction will be inefficient, in which case you may need to use a very high MOI (in the 100s) to get good expression.
The BLOCK-iT™ Adenoviral RNAi Expression System includes the following features designed to enhance its biosafety:
The entire E1 region is deleted in the pAd/BLOCK-iT™-DEST expression vector. Expression of the E1 proteins is required for the expression of the other viral genes (e.g., late genes), and thus viral replication only occurs in cells that express E1 (Graham et al., 1977; Kozarsky and Wilson, 1993; Krougliak and Graham, 1995). This is where the Gateway® Destination cassette is now located. The E3 region has also been deleted.
- Adenovirus produced from the pAd/BLOCK-iT™-DEST expression vector is replication-incompetent in any mammalian cells that do not express the E1a and E1b proteins (Graham et al., 1977; Kozarsky and Wilson, 1993; Krougliak and Graham, 1995).
- Adenovirus does not integrate into the host genome upon transduction. Because the virus is replication-incompetent, the presence of the viral genome is transient and will eventually be diluted out as cell division occurs.
Despite the presence of the safety features discussed above, the adenovirus produced with this System can still pose some biohazardous risk since it can transduce primary human cells. For this reason, we highly recommend that you treat adenoviral stocks generated using this System as Biosafety Level 2 (BL-2) organisms and strictly follow all published guidelines for BL-2. Furthermore, exercise extra caution when creating adenovirus that express shRNA targeting human genes involved in controlling cell division (e.g., tumor suppressor genes) or when producing large-scale preparations of virus (See manual, pg. 11).
For more information about the BL-2 guidelines and adenovirus handling, refer to the document, “Biosafety in Microbiological and Biomedical Laboratories”, 4th Edition, published by the Centers for Disease Control (CDC) (http://www.cdc.gov/od/ohs/biosfty/bmbl4/bmbl4toc.html)
We recommend using One Shot® ccdB Survival T1R chemically competent cells (Cat. No. C751003) for transformation. This strain is resistant to ccdB effects and can support the propagation of plasmids containing the ccdB gene. To maintain integrity for the vector, select for transformants in media containing 50–100 µg/mL ampicillin and 15–30 µg/mL chloramphenicol.
We recommend storing adenoviral expression vectors at –20°C. Due to their relatively large size, we do not recommend storing these vectors at –80°C, as the vector solution will completely freeze, and too many freeze/thaws from –80°C will affect the cloning efficiency.
This is not necessary, as the transfer preserves the orientation of the cassette. However, if you wish to sequence your DEST expression construct, we recommend the following primers:
Primer | Sequence |
pAd forward priming site | 5′-GACTTTGACCGTTTACGTGGAGAC-3′ |
pAd reverse priming site | 5′-CCTTAAGCCACGCCCACACATTTC-3′ |
We use mycoplasma-tested Gibco® FBS (Cat. No. 16000-044) and use the following plasticware for 293A cells:
T-175, Fisher Cat. No. 10-126-13; this is a Falcon flask with 0.2 μm vented plug seal cap.
T-75, Fisher Cat. No. 07-200-68; this is a Costar flask with 0.2 μm vented seal cap.
100 mm plate, Fisher Cat. No. 08-772E; this is a Falcon tissue culture treated polystyrene plate.
We get excellent adherence on these plates under routine cell culture/maintenance conditions (expect cell lysis in 293A cells when making adenovirus).
Any 293-derived cell line or other cell line that expresses the E1 proteins may be used to produce adenovirus. 293A stands for "adherent" because the 293A cells (which are just a single-cell clone of regular 293) tend to adhere and form nice flat monolayers in tissue culture dishes. This is why they work so well for plaque assays. Regular 293 cells will not form the same type of monolayers; they exhibit holes and gaps during growth.
About 10% of transfected cells will make virus. Infected cells ball up and release virus to surrounding cells, which get killed and ball up. Look for plaques created by monolayer areas cleared by detaching, balled-up cells. Squirt off the plaques/cell bodies and harvest.
We generally produce adenoviral stocks in 293A cells using the following optimized transfection guidelines below. The amount of adenovirus produced using these recommended guidelines is approximately 10 mL of crude viral lysate with a titer ranging from 1 x 107 to 1 x 108 plaque-forming units (pfu)/mL.
- Cells: 6-well plate seeded with 5 x 105 cells (293A cells should be plated 24 hours prior to transfection in complete medium, and should be 90–95% confluent on the day of transfection. Make sure that cells are healthy at the time of plating.)
- Transfection mix: 3 µL Lipofectamine® 2000 complexed with 1 μg of PacI-digested pAd-DEST expression plasmid. Note: Verify linearization with PacI prior to transfection. SwaI should also work (if there is a PacI site in the insert). PacI cuts at 34610 and 36684 only; SwaI cuts at 34616 and 36676 only.
- To optimize the transfection, one can titrate Lipofectamine® 2000 and DNA ratios.
- The DNA concentration should be at least 100 ng/µL or more, with A260/A280 ratios (for both the DNA maxi preps and the PacI-digested purified DNA) ratios in the range of 1.8–2.
- Assay: Transfect PacI-cut control DNA into two wells. One well should be stained for lacZ activity at 48 hours posttransfection (this should be 50% blue or better). The other should be transferred to a 100 mm dish to allow the cells to grow up and the plaques to develop.
- Plaques are typically detectable around 10–14 days (sometimes sooner).
Before you can transfect your expression clone into 293A cells, you must expose the left and right viral inverted terminal repeats (ITRs) on the vector to allow proper viral replication and packaging. This also removes bacterial sequences (i.e.,pUC origin and ampicillin resistance gene).
Note: Make sure that your DNA sequence of interest does not contain any PacI restriction sites. If you are unable to use the PacI site, you can use the SwaI site.
Yes, 293A cells are easily transfectable cells, and therefore either reagent can be used and should give high efficiency.
Titering is performed on 293A cell monolayers grown under agar as follows:
1. 6-well plates seeded with 1 x 106 cells per well the day before infection.
2. On the day of infection, to each well add 1 mL each of 10-fold serial dilutions of crude viral lysate ranging from 10–4 to 10–9. Incubate overnight at 37°C.
3. On the following day, remove the infection media and gently overlay the cells with 2 mL Agar Overlay solution per well.
4. After all wells are overlaid, allow the plate to sit level for 15 minutes in the hood at room temperature until the Agar Overlay solidifies.
5. Keep plate in a humidified incubator at 37°C.
6. Two days after the initial overlay, an additional Agar Overlay “feeding” of 1 mL per well is applied on top of the existing overlay. Delaying feeding too long may result in media components being exhausted by cells.
7. Plaques should be visible within 8 days after infection. When plaques are visible to the naked eye (~8 days post-infection), stain plates with the viable stain MTT to make plaque counting easier.
Virus-containing cells can be harvested by squirting the cells off the plate with a 10 mL pipette. Intracellular virus will be released by three freeze/thaws (–80°C/37°C), insoluble material can be removed by centrifugation (3,000 rpm for 15 minutes in a tabletop centrifuge) and the crude viral lysates (CVL) supernatant can be transferred to a fresh tube and stored at –80°C.
Yes, this method is directly scalable to any size of culture dish or roller bottle.
1. Infect 293A cells at 80–90% confluency with virus at an MOI of 3 to 5.
2. Two to three days post-infection, once CPE reached >80%, harvest cells by squirting and release virus by three freeze/thaws.
Please see the following reference for CsCl density gradient centrifugation for concentrating adenovirus:
Engelhardt J F, Yang Y, Stratford-Perricaudet LD et al. (1993) Direct gene transfer of human CFTR into human bronchial epithelia of xenografts with E1-deleted adenoviruses. Nature Genetics 4:27–34.
To amplify the adenoviral stock, we typically infect 3 x 106 293A cells in 10 cm plate (one per adenoviral construct) using 100 µL crude adenoviral stock. Assuming a viral titer of 1 x 107 to 1 x 108 pfu/mL, this generally allows harvest of the desired number adenovirus-containing cells 2–3 days after infection.
- May vary the volume of crude viral stock used to infect cells, if desired. We have used up to 1 mL of crude viral stock.
- If titer of crude viral stock is known, we recommend infecting 293A cells at MOI = 3 to 5.
- Do not amplify the virus more than four times since this increases the chances of generating Recombination Competent Adenovirus (RCA). One should make a high volume stock crude virus and then amplify that in batches once or twice on an as needed basis. You go back to the crude stock when you need more virus.
We recommend aliquoting adenoviral stocks in small working volumes, immediately after production, and storing them at –80°C for long-term storage. Since adenovirus is nonenveloped, viral stocks remain relatively stable and some freezing and thawing of the viral stocks is acceptable. We do not recommend freezing and thawing viral stocks more than 10 times, as loss of viral titer can occur. When stored properly, viral stocks of an appropriate titer should be suitable for use for up to 1 year. After long-term storage, we recommend re-titering your viral stocks before use.
Crude adenovirus titers are generally 1 x 107 to 1 x 108 plaque-forming units (pfu/mL). You can use this stock to infect a new batch of 293A cells to generate a higher titer viral stock (i.e., amplify the virus). Amplification allows production of a viral stock with a titer ranging from 1 x 108 to 1 x 109 pfu/mL. Adenovirus can be concentrated to titers as high as 1 x 1011 pfu/mL using a variety of methods (e.g., CsCl purification).
Please visit our Transfection Support Center to find technical resources, tips and tricks, and troubleshooting information on transfection.
For Research Use Only. Not for use in diagnostic procedures.