Search
RNA Sequencing (RNA-Seq) Methods for NGS
Introduction
Ribonucleic acid (RNA) are polymeric molecules consisting of nucleotides, essential in the coding, decoding, regulation, and expression of genes. In the central dogma of molecular biology, the genetic information of DNA is transcribed into multiple copies of messenger RNA (mRNA), which is then translated into proteins. It is this biological function that allows cells to produce multiple protein molecules from a single gene at a given point in time.
mRNA accounts for only 1-4% of total RNA in a population, and the remainder is generally considered to be noncoding. Noncoding RNA (ncRNA) is defined as RNA that is not translated into protein, and there are many types of ncRNA with a variety of biological functions (1) (Figure 1, Table 1). These types include ribosomal RNA (rRNA), transfer RNA (tRNA), long ncRNA (lncRNA; transcripts longer than 200 nucleotides not translated into protein), and many smaller ncRNAs such as microRNA (miRNA). A DNA sequence encoding an ncRNA is often called an “RNA gene”. The relative amounts of the different types of ncRNAs vary greatly among species and cell types. Most prevalent is rRNA, which typically accounts for 80–95% of the total RNA population. The remainder of ncRNAs are present in much smaller amounts, and thus may require larger samples or enrichment procedures in order to acquire enough material to study.
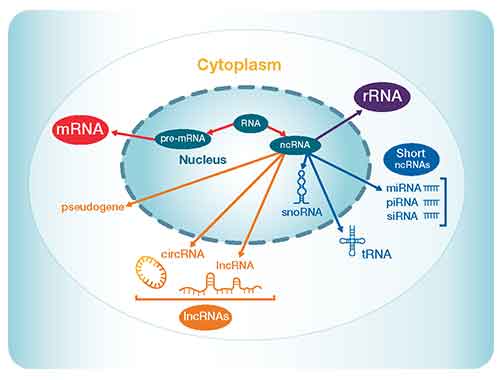
Figure 1. Types of noncoding RNA and their respective locations within a typical eukaryotic cell.
RNA type | Abbreviation | Function |
| Messenger RNA | mRNA | Codes for protein |
| Ribosomal RNA | rRNA | Translation |
| Transfer RNA | tRNA | Translation |
| Small nuclear RNA | snRNA | Splicing and other functions |
| Small nucleolar RNA | snoRNA | Nucleotide modification of RNAs |
| Small Cajal body-specific RNA | scaRNA | Type of snoRNA; nucleotide modification of RNAs |
| Long noncoding RNA | lncRNA | Regulation of gene transcription; epigenetic regulation |
| MicroRNA | miRNA | Gene regulation |
| Piwi-interacting RNA | piRNA | Transposon defense; maybe other functions |
| Small interfering RNA | siRNA | Gene regulation |
Table 1. Types of RNA within a typical eukaryotic cell
What is the transcriptome and RNA-Seq?
The term transcriptome is another way to refer to the total RNA, whether from a single cell or a population of cells. It can also refer to the total mRNA, with a focus on gene expression and what is being specifically coded for proteins. It is clear that understanding the transcriptome has important implications in basic biological discovery, including understanding how gene expression changes on a temporal scale due to external factors, simply by measuring the abundance of a gene-specific transcript. Gene expression analysis can be used to monitor the response of a gene to a treatment with a compound or drug of interest, under a defined set of conditions. Gene expression studies can also involve looking at profiles or patterns of expression of several genes.
RNA sequencing (RNA-Seq) leverages the advantages of next-generation sequencing (NGS) to detect and quantify RNA in a biological sample at a given point in time.
The advantages of RNA-Seq are as follows:
- High throughput sequencing data from NGS enables researchers to study the entire transcriptome with hypothesis-free experimental designs, without the need for probes
- NGS provides broad dynamic range and sensitivity to better detect differentially expressed genes with quantifiable data. One can find the proverbial needle in the haystack within the transcriptome using NGS.
- RNA-Seq is capable of detecting and identifying novel variants, such as alternative splice sites and novel isoforms (see Table 2)
Because of these advantages, researchers have rapidly adopted RNA-Seq as a general method to address this complex landscape. There are now different techniques, from whole transcriptome sequencing to targeted RNA sequencing, that each have their advantages and disadvantages, and which technique to use depends on a researcher’s specific needs.
RNA sequencing method | Description and benefits |
| Total RNA Whole transcriptome | Whole transcriptome analysis to examine coding and noncoding RNA simultaneously; suitable for novel discovery. More throughput intensive to achieve high enough coverage for discovery. Potential inefficiencies and bias due to different sequencing lengths. |
| mRNA sequencing | Poly(A) selection to sequence all messenger RNA for gene expression analysis; able to identify novel and known content |
| smRNA sequencing | Isolation of small RNA to focus study on noncoding RNA to identify novel and known content such as microRNA (miRNA) |
| Targeted RNA sequencing | Sequencing specific transcripts of interest to focus efforts and lower cost to analyze specific genes of interest. Can be used for many sample types, including degraded samples from FFPE. |
Table 2. General RNA sequencing methods
RNA sequencing experimental considerations and workflow
Experimental considerations
As previously noted, various techniques have been developed to best utilize NGS and enable deep sequencing studies of gene expression and novel RNA discovery. Which general approach (Table 2) and experimental design to use is dependent on a variety of parameters:
- Area of research—Is the interest in coding or noncoding RNA, or perhaps both? Are you studying a specific set of genes? Are the experiments for novel discovery, or sequencing known features?
- Sample source—What type of tissue is being studied? Is the sample degraded due to sample source, such as from formalin-fixed paraffin embedded (FFPE) tissue?
- Time dependence—Given RNA and gene expression varies over time, are you performing time course experiments to observe changes? How does this effect the number of samples you are going to sequence? What are the cost and workflow considerations?
- Coverage—What sequencing coverage is required for your experiment, whether to look at expression or for novel smRNA discovery? How does this affect cost and workflow considerations? (Please see the article The Importance of Coverage and Throughput.)
RNA sequencing general workflow
RNA samples are typically fragmented to a specific size range if one is studying the whole transcriptome or performing mRNA sequencing. This is due to sequencing read length limitations on current NGS platforms. RNA fragmentation can be done in a variety of ways, such as with divalent cation solutions or an enzymatic approach such as using RNase III. Alternatively, one could fragment full-length cDNA prior to entering the NGS library construction workflow.
Regardless of the method used for the RNA study, RNA-Seq requires reverse transcription to synthesize complementary DNA (cDNA) for analysis on current NGS instruments. Maintaining RNA strand information is important to accurately identify novel species and measure sense RNA expression (2). It is estimated that in the human genome, nearly 20% of genes are overlapping and are transcribed from opposite strands (3). Thus, it is important to use the correct approach and adapters to capture the directionality of RNA in the resulting cDNA.
Depletion of ribosomal RNA before fragmentation is also frequently performed given it is the most abundant RNA and typically of little interest. By removing rRNA, sequencing reads and resulting throughput is focused on your transcripts of interest. One approach to remove rRNA is to use sequence-specific probes, whether in the form of biotinylated DNA or locked nucleic acid (LNA) probes, which will hybridize to the targets and subsequently be removed from the wanted material using streptavidin beads. An enzymatic approach using Ribonuclease H (RNAse H) to selectively cleave RNA/DNA substrates can also be used, but this requires a set of probes unique for different species to bind to the target rRNA.
Targeted RNA sequencing workflow
Targeted RNA sequencing provides a simple and cost-effective alternative compared to whole transcriptome and mRNA sequencing, which uses a fragmentation workflow. Through targeted cDNA amplification, targeted RNA sequencing can be used to focus on specific transcripts of interest and bypass the need for rRNA depletion. A targeted approach can also be performed with limited RNA samples, whether low input amounts or highly degraded samples such as RNA material from formalin-fixed paraffin embedded (FFPE) tissue. One downstream benefit is the actual data analysis workflow, which is now streamlined to examine specific transcripts.
Please see the article Targeted Sequencing Approaches for NGS to learn how hybridization capture and amplicon-based methods are used to help focus sequencing on specific genetic information.
References
- Palazzo AF, Lee ES. Front Genet 6:2 (2015)
- Hrdlickova R, et al. WIREs RNA 8:e1364 (2017)
- Zhao S, et al. BMC Genomics 16(1):675 (2015)
Additional resources
For Research Use Only. Not for use in diagnostic procedures.