Search
When calculating the results of your real-time PCR (qPCR) experiment, you can use either absolute or relative quantification.
| Absolute quantification (digital PCR method) | Absolute quantification(standard curve method) | Relative quantification | |
|---|---|---|---|
| Overview | In absolute quantification using digital PCR, no known standards are needed. The target of interest can be directly quantified with precision determined by number of digital PCR replicates. | In absolute quantification using the standard curve method, you quantitate unknowns based on a known quantity. First you create a standard curve; then you compare unknowns to the standard curve and extrapolate a value. | In relative quantification, you analyze changes in gene expression in a given sample relative to another reference sample (such as an untreated control sample). |
| Example | Quantify copies of rare allele present in heterogeneous mixtures. Count the number of cell equivalents in sample by targeting genomic DNA. Determine absolute number of viral copies present in a given sample without reference to a standard. | Correlating viral copy number with a disease state. | Measuring gene expression in response to a drug. In this example, you would compare the level of gene expression of a particular gene of interest in a chemically-treated sample relative to the level of gene expression in an untreated sample. |
Digital PCR works by partitioning a sample into many individual real-time PCR reactions; some portion of these reactions contain the target molecule (positive), while others do not (negative). Following PCR analysis, the fraction of negative answers is used to generate an absolute answer for the exact number of target molecules in the sample, without reference to standards or endogenous controls.
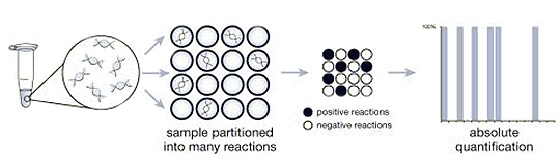
Figure 1: Digital PCR uses the ratio of positive (white) to negative (black) PCR reactions to count the number of target molecules.
The standard curve method for absolute quantification is similar to the standard curve method for relative quantification, except the absolute quantities of the standards must first be known by some independent means.
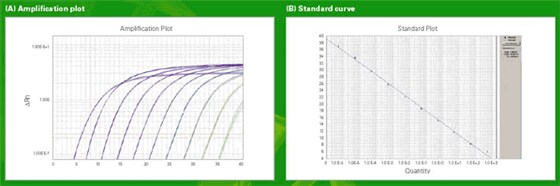
Figure 2: Amplification plot and standard curve for absolute quantification
Critical guidelines
The guidelines below are critical for proper use of the standard curve method for absolute quantification:
- It is important that the DNA or RNA be a single, pure species. For example, plasmid DNA prepared from E. coli often is contaminated with RNA, which increases the A260 measurement and inflates the copy number determined for the plasmid.
- Accurate pipetting is required because the standards must be diluted over several orders of magnitude. Plasmid DNA or in vitro transcribed RNA must be concentrated in order to measure an accurate A260 value. This concentrated DNA or RNA must then be diluted 106–1012 -fold to be at a concentration similar to the target in biological samples.
- The stability of the diluted standards must be considered, especially for RNA. Divide diluted standards into small aliquots, store at –80 °C, and thaw only once before use.
It is generally not possible to use DNA as a standard for absolute quantification of RNA because there is no control for the efficiency of the reverse transcription step.
Standards
The absolute quantities of the standards must first be known by some independent means. Plasmid DNA and in vitro transcribed RNA are commonly used to prepare absolute standards. Concentration is measured by A260 and converted to the number of copies using the molecular weight of the DNA or RNA.
Calculation methods for relative quantification
Relative quantification can be performed with data from all real-time PCR instruments. The calculation methods used for relative quantification are:
- Standard curve method
- Comparative CT method
Figure 3: Relative quantification
Overview | ||
| Digital PCR method | Standard curve method | Comparative CT method |
|---|---|---|
| Nucleic acid quantification utilizes theory of limiting sample dilution that is spread across many technical replicate PCR reactions. Absolute quantification is determined by ratio of number of negative versus total reactions. This type of analysis is different from Ct and delta Ct comparisons, and instead allows each assayed target to be quantified independently without the need for reference standards. | It is easy to prepare standard curves for relative quantification because quantity is expressed relative to some basis sample (called a calibrator), such as an untreated control. For all experimental samples, you determine target quantity from the standard curve and divide by the target quantity of the calibrator. Thus, the calibrator becomes the 1× sample, and all other quantities are expressed as an n-fold difference relative to the calibrator. | This method compares the Ct value of one target gene to another (using the formula: 2ΔΔCT)—for example, an internal control or reference gene (e.g., housekeeping gene)—in a single sample. |
Advantages | ||
| No need to rely on references or standards. Desired precision can be achieved by increasing total number of PCR replicates. Highly tolerant to inhibitors. Capable of analyzing complex mixtures. Unlike traditional qPCR, digital PCR provides a linear response to the number of copies present to allow for small-fold change differences to be detected | Running the target and endogenous control amplifications in separate tubes and using the standard curve method of analysis requires the least amount of optimization and validation. | You don't need a standard curve and can increase throughput because wells no longer need to be used for the standard curve samples. This also eliminates dilution errors made in creating the standard curve samples. You can amplify the target and endogenous control in the same tube, increasing throughput and reducing pipetting errors. When RNA is the template, performing amplification in the same tube provides some normalization against variables such as RNA integrity and reverse transcription efficiencies. |
Experimental validation | ||
| Validated digital PCR results with side-by-side comparison to a well-characterized sample with known copy number. | See Advantages above. | You have to run a validation experiment to show that the efficiencies of the target and endogenous control amplifications are approximately equal. |
Critical guidelines | ||
| It is important to use low-binding plastics as much as possible throughout experimental set-up. Since digital PCR emphasizes assaying limiting dilution, any sample that sticks to intermediate set-up equipment will be lost and skew results. We recommend using low binding 2.0 mL tubes for dilutions and low-retention pipette tips. It is important to know the digital area of the desired sample to be tested. If unknown, consult user guide for help in determining copy number of known species (gDNA) or perform a preliminary screening experiment with multiple dilutions of sample/assay combination to determine optimal digital concentration to ensure meaningful data attained. Sample should not be kept at low concentration for extended periods of time, nor exposed to excessive freeze-thawing. Carriers have not been shown to be as important to reproducibility as much as using non-stick plastics for experimental set-up. Careful planning of dilutions is desired to minimize variability due to dilution scheme. | It is important that stock RNA or DNA be accurately diluted, but the units used to express this dilution are irrelevant. If two-fold dilutions of a total RNA preparation from a control cell line are used to construct a standard curve, the units could be the dilution values 1, 0.5, 0.25, 0.125, and so on. By using the same stock RNA or DNA to prepare standard curves for multiple plates, the relative quantities determined can be compared across the plates. You can use a DNA standard curve for relative quantification of RNA. Doing this assumes that the reverse transcription efficiency of the target is the same in all samples, but the exact value of this efficiency need not be known. For quantification normalized to an endogenous control, standard curves are prepared for both the target and the endogenous reference. For each experimental sample, the amount of target and endogenous reference is determined from the appropriate standard curve. Then, the target amount is divided by the endogenous reference amount to obtain a normalized target value. Again, one of the experimental samples is the calibrator, or 1× sample. Each of the normalized target values is divided by the calibrator normalized target value to generate the relative expression levels. | For the comparative CT method to be valid, the efficiency of the target amplification (your gene of interest) and the efficiency of the reference amplification (your endogenous control) must be approximately equal. |
Endogenous control | ||
| Digital PCR does not rely on the presence of endogenous controls for reference measurements. | Amplification of an endogenous control may be performed to standardize the amount of sample RNA or DNA added to a reaction. For the quantification of gene expression, researchers have used ß-actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ribosomal RNA (rRNA), or other RNAs as an endogenous control. | |
Standards | ||
| Because digital PCR uses the fraction of negative to total replicates to determine an absolute count of molecules, no standards are required. | Because the sample quantity is divided by the calibrator quantity, the unit from the standard curve drops out. Thus, all that is required of the standards is that their relative dilutions be known. For relative quantification, this means any stock RNA or DNA containing the appropriate target can be used to prepare standards. |
For Research Use Only. Not for use in diagnostic procedures.