Search
Overview of Protein–Nucleic Acid Interactions
In the late 19th century, scientists microscopically observed the association of proteins with DNA strands. Since then, researchers have used a variety of in vitro and in vivo assays to demonstrate that proteins interact with DNA and RNA to influence the structure and function of the corresponding nucleic acid. Elucidating the roles that protein–nucleic acid complexes play in the regulation of transcription, translation, DNA replication, repair and recombination, RNA processing and translocation continues to revolutionize our understanding of cell biology, normal cell development and the mechanisms of disease. This article provides an introduction to some of the key methods used study protein–nucleic acid interactions.
Introduction to protein–nucleic acid interactions
Proteins interact with DNA and RNA through similar physical forces, which include electrostatic interactions (salt bridges), dipolar interactions (hydrogen bonding, H-bonds), entropic effects (hydrophobic interactions) and dispersion forces (base stacking). These forces contribute in varying degrees to proteins binding in a sequence-specific (tight) or non–sequence-specific (loose) manner. For example, specific protein–DNA interactions are commonly mediated by an α-helix motif in the protein that inserts itself into the major groove of the DNA, recognizing and interacting with a specific base sequence through H-bonds and salt bridges. In addition, the affinity and specificity of a particular protein–nucleic acid interaction can be increased through protein oligomerization or multi-protein complex formation (e.g., GCN4, glucocorticoid receptor, transcription initiation complexes, mRNA splicing complexes, RISC, etc.). The secondary and tertiary structure formed by nucleic acid sequences (especially in RNA) provides an important additional mechanism by which proteins recognize and bind particular nucleic acid sequences.
Application note: Analysis of Androgen-dependent and -independent Regulation of Transcriptional Activity
This application note describes the use of chromatin immunoprecipitation (ChIP) assay to monitor the interaction between the androgen receptor (AR) and androgen response elements (AREs) in the DNA. The LNcaP prostate cancer cell line was exposed to testosterone and the Thermo Scientific Pierce Magnetic ChIP Kit was used with an anti-AR antibody followed by qPCR. The figure to the left shows changes in AR binding to known AREs (PSA, CDKN1A, FKBP5, TMPRSS2 and IGF-1), with a 300-fold change to the FKBP5 ARE 30 minutes after treatment. This kit allows for the efficient isolation of chromatin-bound DNA by immunoprecipitation in about 8 hours with as few as 10,000 cells.

Protein Interactions Handbook
Our 72-page Protein Interaction Technical Handbook provides protocols and technical and product information to help maximize results for protein interaction studies. The handbook provides background, helpful hints and troubleshooting advice for immunoprecipitation and co-immunoprecipitation assays, pull-down assays, far-western blotting and crosslinking. The handbook also features an expanded section on methods to study protein–nucleic acid interactions, including ChIP, EMSA, and RNA EMSA. The handbook is an essential resource for any laboratory studying protein interactions.
Contents include: Introduction to protein interactions, Co-immunoprecipitation assays, Pull-down assays, Far-western blotting, Protein interaction mapping, Yeast two-hybrid reporter assays, Electrophoretic mobility shift assays [EMSA], Chromatin immunoprecipitation assays (ChIP), Protein–nucleic acid conjugates, and more.

Nucleic acid binding domains
The DNA- or RNA-binding function of a protein is localized in discrete conserved domains within its tertiary structure. An individual protein can have multiple repeats of the same nucleic acid binding domain or can have several different domains found within its structure. The identity of the individual domains and their relative arrangement are functionally important within the protein. Several common DNA binding domains include zinc fingers, helix-turn-helix, helix-loop-helix, winged helix and leucine zipper. RNA-binding specificity and function are constituted by zinc finger, KH, S1, PAZ, PUF, PIWI and RRM (RNA recognition motif) domains. Multiple nucleic acid binding domains with a single protein can increase specificity and affinity of the protein for certain target nucleic acid sequences, mediate a change in the topology of the target nucleic acid, properly position other nucleic acid sequences for recognition or regulate the activity of enzymatic domains within the binding protein.
Complex interactions
Proteins can bind directly to the nucleic acid or indirectly through other bound proteins, effectively creating a hierarchy of interactions. The strength of these interactions influence which assays or approaches are best for studying complex assembly. Some of these interactions are transient and require stabilization through chemical crosslinking prior to isolation of the complexes. Understanding how proteins interact with nucleic acids, determining what proteins are present in these protein-nucleic acid complexes and identifying the nucleic acid sequence/structure required to assemble these complexes are vital to understanding the role these complexes play in regulating cellular processes.
Protein–DNA interactions
The common DNA-binding domains, helix-turn-helix and zinc finger domains, are incorporated within numerous DNA-binding proteins expressed in the cell. Specificity is derived from higher order interactions involving nucleoprotein complexes. These DNA-binding protein complexes find their target by “sliding” along the genomic DNA until their specific DNA-docking site is discovered. The binding of protein to DNA controls the structure of genomic DNA (chromatin), RNA transcription, and DNA repair mechanisms. The following example illustrates how Invitrogen Dynabeads magnetic beads may be used to recover proteins that bind to nucleic acids.
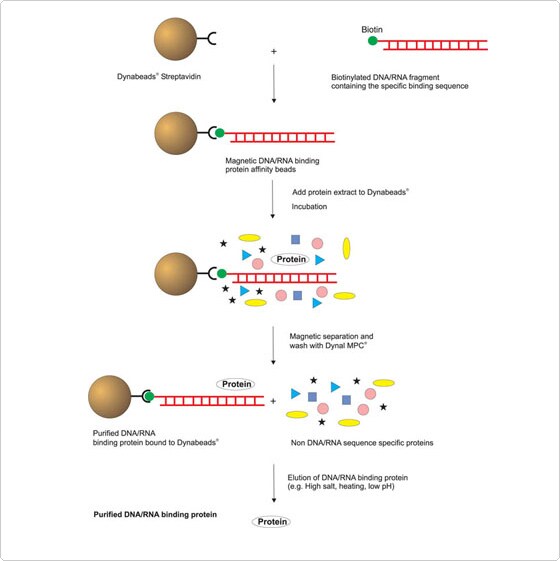
Chromosomes
A major function of protein–DNA interactions is to manage the extensive length of the genetic material contained in each cell. Chromosomes have evolved to package, store and move DNA throughout the cell, but they also play a role in transcriptional regulation. Chromosome remodeling allows selective portions of a chromosome to be unraveled so that the DNA is available for gene transcription, or to remain tightly packaged so that transcription of the encoded genes is completely silenced.
Transcriptional regulation
Once unraveled, genomic DNA can be transcribed; however, not all of the DNA sequence codes for proteins. Only genes are transcribed to produce RNA, and the sequences between the genes (and within) serve to regulate transcription through protein binding. These sequences are important for transcriptional control, and they contain promoters, enhancers, insulators and spacers. Enhancer sequences, which can be many kilobases away from the gene start site, bind proteins and act as beacons to attract the transcriptional machinery. Transcription of a gene is initiated when transcription factor proteins bind to specific DNA promoter sequences located immediately adjacent to the gene transcription start site. This interaction is facilitated through the DNA-binding domain(s) of the transcription factor. Through protein interactions, the trans-activation domain of the transcription factor facilitates binding and localization of the RNA polymerase II holoenzyme to the gene promoter in order to initiate production of a messenger RNA (mRNA)
Protein–RNA interactions
Proteins interact with RNA in order to splice, protect, translate or degrade the message. The first interaction occurs just after transcriptional initiation, when the complement to the promoter sequence is cleaved out of the mRNA and the capping machinery incorporates a "GpppN" cap at the 5' end of the mRNA. This results in recruitment of elongation factors that regulate the reset of mRNA transcription. Elongation is followed by 3'-end processing and splicing, resulting in a mature RNA transcript that is exported to the cytoplasm for translation. All of these processes require significant protein–RNA interactions and are highly regulated and complex. Many of the regulatory elements for this process reside in noncoding 3' and 5' untranslated regions (UTRs) of the mRNA. However, regulatory microRNAs (miRNAs) also occur in coding regions of introns, as well as exons, noncoding genes and repetitive elements. In recent years, increased emphasis has been placed on the importance of these noncoding RNA sequences and their roles in cellular regulation and disease states. However, tools for the study of critical protein–RNA interactions have been limited. The data shown in this example were generated using an RNA–protein pull-down experiment using the Thermo Scientific Pierce Magnetic RNA-Protein Pull-Down Kit.
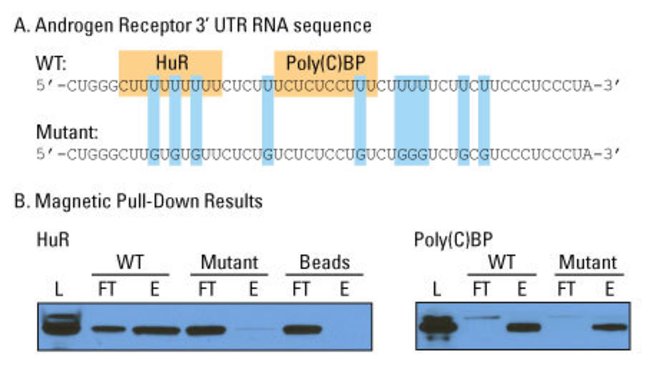
RNA–protein pull-down experiment. The binding of wild-type and mutant androgen receptor 3’-UTR RNA to proteins HuR and Poly(C)BP was assayed using the Pierce Magnetic RNA-Protein Pull-Down Kit. Samples were normalized by volume. L = lysate load, FT = flow-through, E = eluate. This kit provides a robust method to enrich protein–RNA interactions.
5' and 3' UTRs
The UTR regions of mRNA contain sequence elements that recruit RNA-binding proteins for post-transcriptional regulation and protein translation. In addition, these elements promote transcript stability or degradation, and they can direct subcellular localization of the RNA. These RNA regulatory elements vary in length but rely on both primary and secondary structure for protein binding. For example, iron regulation in the cell is a tightly regulated process where a protein–RNA interaction is key to maintaining iron homeostasis. Target genes, such as the iron storage protein ferritin or the transferrin receptor, contain a small (~28 nucleotides) consensus iron responsive element (IRE) in their respective 5' or 3' UTR. The iron responsive protein (IRP) responds to cellular iron status by binding the IRE element. Under iron-starved conditions, IRP remains bound to the IRE element to suppress translation of iron storage proteins. Under iron-rich conditions, IRE binding activity of IRP is lost and iron storage proteins are translated. Many of these RNA consensus elements have been identified and classified into different families based on sequence and function. The 3' UTR also contains recognition elements for miRNA, which are responsible for repressing protein translation of the coding mRNA.
MicroRNA
MicroRNAs (miRNAs) are a large and ubiquitous class of noncoding RNAs that regulate post-transcriptional silencing of target mRNA. Over 700 miRNAs have been identified in the human genome. MicroRNAs have binding recognition sequences in 57.8% of human mRNAs, with 72% containing of those mRNAs having multiple miRNA recognition sites. The miRNA begins as a 70–100 nucleotide transcribed RNA (pre-miRNA) containing a 6–8 nucleotide seed region at the 5' end for mRNA binding. The miRNA is then cleaved by DROSHA, a nuclear endoribonuclease III. The pre-miRNA then associates with double-stranded RNA-binding proteins and is actively exported to the cytoplasm, dependent on Exportin 5 and Ran GTPase. The pre-miRNA is then further processed in a ribonucleoprotein (RNP) complex consisting of Argonaute proteins and Dicer (endoribonuclease III), which cleaves the pre-miRNA into the mature 19–22 nucleotide miRNA. The miRNA-Argonaute complex then binds to target genes and recruits additional unidentified proteins for regulation of target genes.
mRNA regulation
In most cases, the mRNA regulation results in repression of translation through mRNA degradation, deadenylation or storage in cytoplasmic mRNA processing bodies (P-bodies), but mRNA translation may also be up-regulated. Several models of mRNA repression and degradation have been proposed, but a single accepted model has not yet been adopted. MicroRNA research is rapidly expanding and key protein–RNA interactions are being investigated to further understand the role of miRNA in cell growth, differentiation and carcinogenesis.
Recommended reading
- Hendrickson W (1985) BioTechniques 3:346–354.
- Evertts AG et al. (2010) Modern approaches for investigating epigenetic signaling pathways. J Appl Physiol Jan 28 ePub ahead of print.
- Georges AB et al. (2010) Generic binding sites, generic DNA-binding domains: Where does specific promoter recognition come from? FASEB Journal 24:346–356.
- Lunde BM et al. (2007) RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 8:479–490.
For Research Use Only. Not for use in diagnostic procedures.