Search
Biological Discovery Is a Click Away
Click chemistry uses biologically unique moieties to label and detect molecules of interest. Click reactions have several advantages: the reaction between the labeling and detection moieties is simple and efficient; no extreme temperatures or solvents are required; and the reaction product is stable. Perhaps most importantly, the components of the click reaction are bioinert—the label and detection tags react selectively with one another without complications from unwanted side reactions [1]. This selectivity means that click chemistry–based labeling and detection protocols can be applied to complex biological samples with unprecedented sensitivity, thanks to extremely low background levels.The Click-iT® assays are based on a two-step reaction involving the copper-catalyzed formation of a 1,2,3-triazole linkage between an azide and an alkyne. The azide and alkyne moieties can be used interchangeably: either one can be used to tag the molecule of interest while the other is used for subsequent detection. The label is small enough that tagged molecules (e.g., nucleotides, sugars, and amino acids) are acceptable substrates for the enzymes that assemble these building blocks into biopolymers. With its small “footprint,” the Click-iT® detection molecule can easily penetrate complex samples, including intact (supercoiled) DNA, with only mild permeabilization required. Click chemistry serves as a useful alternative when methods such as direct labeling or antibody-based detection fall short.
Accurate Detection of Cell Proliferation in Single-Pulse or Dual-Pulse Experiments
Click-iT® EdU assays are used to accurately measure proliferation through the use of a modified nucleoside, EdU (5-ethynyl-2´-deoxyuridine), which is incorporated during DNA synthesis. Although Click-iT® EdU technology has only been available since 2007, its use has already been demonstrated in a wide variety of species, including plants, bacteria, yeasts, and a broad spectrum of animals (Figure 1) [2–9].
In applications where only a single pulse is required, Click-iT® EdU assays provide not only a greatly truncated protocol compared to BrdU-based proliferation assays by eliminating the DNA denaturation step, but also an extremely bright signal without the incubations required by secondary detection methods for signal amplification. For dual-pulse applications, the use of EdU as one of the nucleoside analogs greatly simplifies the procedure, as there is no reactivity between the Click-iT® azide detection reagent and the incorporated BrdU. Moreover, several anti-BrdU antibodies, including the MoBU-1 clone, do not cross-react with EdU (Figure 2). The use of BrdU and EdU together have allowed temporal characterization of S-phase cells to investigate the relationship between cell-cycle length and neurogenesis [10].
More Information
- Learn More About Click-iT® Detection Assays
Download This Article
Get a copy of this article as it appears in the print version of BioProbes 65.
Quick Product View
See a complete listing of the products discussed in this article.
View Products

 | Figure 1. Detection of DNA synthesis in the flatworm Flagellophora cf. apelti. Cells were exposed to EdU (100 μM in seawater) for 10 hr. Following fixation and permeabilization, EdU was detected with the Click-iT® EdU Alexa Fluor® 488 Imaging Kit (green fluorescence). Phospho-H3 was detected using a rabbit primary antibody followed by an Alexa Fluor® 568 dye–labeled goat anti–rabbit IgG antibody (red fluorescence). Image submitted by Julian Smith, Department of Biology, Winthrop University, USA. |
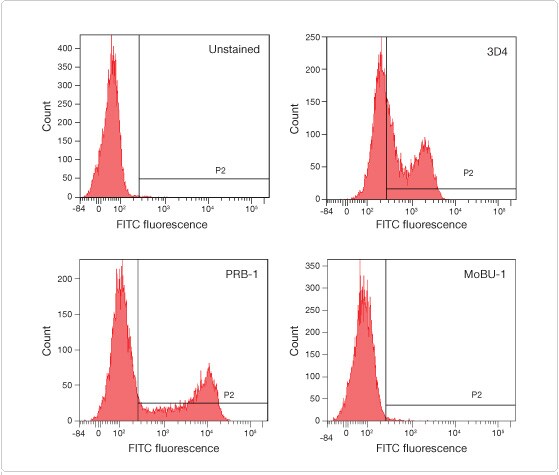
Figure 2. The anti-BrdU antibody clone MoBU-1 does not react with EdU. Jurkat T cells were treated with 10 μM EdU for 1 hr, the cells were fixed in ethanol, and an acid denaturation method was used before labeling with FITC conjugates of the most commonly used anti-BrdU clones: 3D4, PRB-1, and MoBU-1. Cells were analyzed with a BD™ LSR II flow cytometer using a 488 nm excitation laser, and fluorescence was collected with a 530/30 bandpass filter. Of the clones tested, only clone MoBU-1 did not react with EdU.
Mitochondrial DNA Synthesis Exposed
Changes in mitochondrial DNA (mtDNA) copy number are not only important for normal biological processes but also implicated in drug toxicity, aging, and a wide range of diseases, including cancer, diabetes, and neurodegeneration. Methods used to study the regulation of nuclear DNA replication are, in theory, applicable to mtDNA applications but have faced challenges in practice. Visualizing nascent mtDNA using the Click-iT® EdU assay requires signal amplification. Tyramide Signal Amplification (TSA®) technology (PerkinElmer) is compatible with the Click-iT® EdU assay and has been reported to increase detection sensitivity up to 100-fold compared to conventional avidin–biotinylated enzyme complex (ABC) procedures [11]. By combining the streamlined Click-iT® EdU assay with ultrasensitive TSA® detection, a superior method is established to explore the biology of mtDNA replication and turnover [12] (Figure 3), and for toxicological profiling of drugs that inhibit mtDNA synthesis.
 | Figure 3. mtDNA replication in dissociated dorsal root ganglion neurons. Neurons plated on glass coverslips were incubated with 10 μM EdU. Following fixation and permeabilization, endogenous peroxidase activity was blocked and EdU was detected with Oregon Green® 488 azide. The signal was amplified using HRP-conjugated rabbit antibody against Oregon Green® 488 and Alexa Fluor® 488 tyramide (green) from TSA® Kit #12. The pan-neuronal marker αTuj1 was detected with a mouse primary antibody and visualized with an Alexa Fluor® 594 goat anti-mouse secondary antibody (red). Nuclei were stained with DAPI (blue). Image contributed by Stephen I. Lentz and Eva L. Feldman, Departments of Internal Medicine and Neurology, University of Michigan. |
Imaging Global RNA Synthesis
Click-iT® RNA assays are ideal for imaging global RNA synthesis in multiplex analyses using traditional fluorescence microscopy or high-content screening (HCS). The small size of the alkyne moiety (MW ~25) of EU (5-ethynyl uridine) enables efficient incorporation by RNA polymerases without any apparent changes to the RNA levels of several housekeeping genes [5]. The multiplexing capability of the assays makes them ideal for toxicological profiling or interrogation of disease models using high-content imaging platforms (Figure 4). The Click-iT® RNA Imaging and Click-iT® RNA HCS kits also provide exciting new alternatives to antibody-based BrU or BrUTP assays for studying viral RNA synthesis in infected cells (Figure 5).
 | Figure 4. Dose response for actinomycin D in HeLa cells using the Click-iT® RNA Assay. HeLa cells were treated with the indicated amounts of actinomycin D for 18 hr, followed by a 1 hr incubation with 5-ethynyl uridine (EU). Cells were then fixed and permeabilized, and EU incorporated into newly synthesized RNA was detected using green-fluorescent Alexa Fluor® 488 azide. Quantitative analysis was performed using the Cellomics® ArrayScan® VTI and Compartmental Analysis Bioapplication (Thermo Scientific). |
 | Figure 5. Subcellular localization of RNA in Vero cells. Cells were pretreated with 2 μM actinomycin D to inhibit host cell transcription, then infected with Tacaribe virus. Infected cells were incubated with 2 mM EU for 1 hr, followed by cold methanol fixation and permeabilization with Triton® X-100. Nascent RNA (green) was detected with the Click-iT® RNA Alexa Fluor® 488 Imaging Kit. Viral nucleoprotein was detected with a directly labeled Alexa Fluor® 594 monoclonal antibody (red). Colocalization of EU and virus nucleoprotein indicates transcription sites in the host cells (yellow). |
Study the Kinetics of Transcription and RNA Turnover
Until now, the only assay that has been widely used to generate nascent RNA for transcriptional profiling is the cumbersome nuclear run-on assay. Because transcription is measured in isolated nuclei in vitro, a major limitation of this method is that the procedure is cell-invasive and subject to experimental bias. Moreover, nuclear run-on does not allow for subsequent capture of nascent RNA. The Click-iT® Nascent RNA Capture Kit offers a simple, efficient method of capturing newly synthesized RNA for higher-resolution studies of the transcriptome that is not cell-invasive, nor does it require radioactive nucleoside analogs. The Click-iT® Nascent RNA Capture Kit enables simultaneous analysis of short-term changes in RNA synthesis and decay, and their impact on overall cellular transcript levels. To investigate the kinetics of RNA synthesis, the duration of EU treatment can be varied, followed by nascent RNA capture. mRNA turnover can then be estimated by dividing the Ct value for the nascent RNA by the Ct value for total RNA. For analysis of RNA decay, a traditional pulse-chase experiment can be performed to determine the loss of labeled RNA (Figure 6).
 | Figure 6. The Click-iT® Nascent RNA Capture Kit can be used to analyze the kinetics of RNA decay. Jurkat cells were pulsed with EU for 24 hr. Total RNA was isolated and subjected to nascent RNA capture, and real-time PCR analysis was performed. Fold loss of RNA was calculated as follows: [Ct pulse] – [Ct pulse + chase] = ΔCt fold change = 2–ΔCt. |
The Next Generation in Protein Profiling
Several recent publications [13–15] reveal that click chemistry–based tools are able to reliably and sensitively detect the presence of post-translational modifications (PTMs), delivering detection data in hours or days as opposed to the weeks or months required with radiolabeling techniques. We offer several click-modified reagents for in-depth protein analysis (Table 1). The azide- or alkyne-containing biomolecule is fed to cells or animals and becomes actively incorporated into proteins, allowing radioisotope-free detection of key PTMs and nascent protein synthesis (Figure 7).
In addition to our growing selection of labeled biomolecules, the Click-iT® O-GlcNAc Enzymatic Labeling System can be used for in vitro labeling of O-GlcNAc–modified glycoproteins. Once labeled, the modified protein is detected with the corresponding alkyne-containing dye or hapten using either the Click-iT® Cell Reaction Buffer Kit or the Click-iT® Protein Reaction Buffer Kit. With the Click-iT® Cell Reaction Buffer Kit, cells can be analyzed by fluorescence microscopy, flow cytometry, or high-content imaging and analysis. With the Click-iT® Protein Reaction Buffer Kit, proteins are compatible with common analyses including downstream LC-MS/MS and MALDI MS analysis, and detection sensitivity in 1D gels and western blots is in the low femtomole range.
Table 1. Azide- and alkyne-modified biomolecules for analysis of protein synthesis and posttranslational modifications.
| Biomolecule | Azide or Alkyne | Use * |
|---|---|---|
| L-Azidohomoalanine | Azide | Monitor nascent protein synthesis and turnover |
| L-Homopropargylglycine | Alkyne | |
| Fucose alkyne | Alkyne | Identify fucosylated proteins |
| GalNAz (tetraacetylated N-azidoacetylgalactosamine) | Azide | Identify O-linked glycoproteins including O-GlcNAc |
| GlcNAz (tetraacetylated N-azidoacetylglucosamine) | Azide | Identify O-GlcNAc–modified glycoproteins |
| ManNAz (tetraacetylated N-azidoacetyl-D-mannosamine) | Azide | Identify sialic acid–modified glycoproteins |
| Geranylgeranyl alcohol, azide | Azide | Identify geranylgeranylated proteins |
| Palmitic acid, azide (15-azidopentadecanoic acid) | Azide | Identify protein fatty acylation |
| Myristic acid, azide (12-azidododecanoic acid) | Azide | |
| * Also available, the Click-iT® Protein Enrichment Kit (C10416) for click chemistry capture of azide-modified proteins and subsequent mass spectrometry analysis. | ||

Figure 7. Comparison of 35S-methionine and Click-iT® AHA assay sensitivity. Cortical neurons were metabolically labeled with either Click-iT® AHA (L-azidohomoalanine) or 35S-methionine (35S-met) in a time series for 15–90 min. (Left) Newly synthesized proteins labeled with Click-iT® AHA were detected with the Click-iT® TAMRA Protein Analysis Kit and visualized using 532 nm excitation. (Right) Proteins labeled with 35S-met were detected by storage phosphorimaging. The time course for incorporation of Click-iT® AHA vs. 35S-met shows similar increases over a time frame of 15–90 min. The sensitivity of Click-iT® detection is comparable to that of 35S-met; the fluorescence of the TAMRA image was quantified in the two most prominent bands, and the incorporation was linear across the entire time series (data not shown). Data contributed by Pate Skene, Duke University.
Detect DNA Fragmentation With Click-iT® TUNEL Assays
At the final stage of apoptosis, adherent cells often detach from the slide or well before they can be detected. Because cells are lost in this way, you may not be able to detect the DNA fragmentation—a hallmark and the ultimate determinant of apoptosis. Since its introduction in 1992 [16], the most widely used in situ test for studying DNA fragmentation or strand breaks is the terminal deoxynucleotidyl transferase-dUTP nick end labeling (TUNEL) assay, which is based on the incorporation of modified dUTPs by terminal deoxynucleotidyl transferase (TdT) at the 3´-OH ends of fragmented DNA. For a reliable and reproducible TUNEL imaging assay, it is not only vital that the modified nucleotide be an acceptable substrate for TdT, but also that the detection method be sensitive and avoid any additional loss of cells from the sample. The Click-iT® TUNEL Imaging Assays meet both of these criteria. The alkyne-modified dUTP used in the Click-iT® TUNEL assay is rapidly incorporated by TdT, providing sensitive detection that allows you to fix your samples earlier and preserve late-stage apoptotic cells for imaging before they detach.
Making Biodiscovery a Snap
Life Technologies has been the leader in providing click chemistry reagents and assays for use in biological systems. This article highlights some of the applications and a few of the more than 1,400 publications to date that report the utility of click chemistry for biomolecule labeling and detection.
References
- Kolb HC, Finn MG, Sharpless KB (2001) Angew Chem Int Ed Engl 40:2004–2021.
- Dorsett M, Westlund B, Schedl T (2009) Genetics 183:233–247.
- Bando T, Mito T, Maeda Y et al. (2009) Development 136:2235–2245.
- Kaiser CL, Kamien AJ, Shah PA (2009) Laryngoscope 119:1770–1775.
- Salic A (2008) Proc Natl Acad Sci U S A 105:2415–2420.
- Bonaguidi MA, Peng CY, McGuire T et al. (2008) J Neurosci 28:9194–9204.
- Kharas MG, Janes MR, Scarfone VM et al. (2008) J Clin Invest 118:3038–3050.
- McCord AM, Jamal M, Williams ES et al. (2009) Clin Cancer Res 15:5145–5153.
- Momcilovic O, Choi S, Varum S et al. (2009) Stem Cells 27:1822–1835.
- Zeng C, Pan F, Jones LA et al. (2010) Brain Res 1319:21–32.
- Van Heusden J, de Jong P, Ramaekers F et al. (1997) J Histochem Cytochem 45:315–320.
- Lentz SI, Edwards JL, Backus C et al. (2010) J Histochem Cytochem 58:207–218.
- Martin DD, Vilas GL, Prescher JA et al. (2008) FASEB J 22:797–806.
- Kostiuk MA, Corvi MM, Keller BO et al. (2008) FASEB J 22:721–732.
- Wang Z, Park K, Comer F et al. (2009) Diabetes 58:309–317.
- Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) J Cell Biol 119:493–501.
For Research Use Only. Not intended for any animal or human therapeutic or diagnostic use.
For Research Use Only. Not for use in diagnostic procedures.