Search
Fluorescent Probes for Super-Resolution Imaging Technologies
Ever try to drive to the grocery store with a foggy windshield? Or watch your favorite sports team in full action without your prescription glasses? Knowing what is possible to see makes us less accepting of having less-than-perfect vision. With the advent of super-resolution microscopy (SRM), also known as nanoscopy, the limitations of diffraction-limited fluorescence microscopy are becoming clear. SRM technology, which depends on optimized hardware, software, and fluorescent probes, may change our expectations for cellular imaging.SRM: Breaking the Diffraction Barrier
SRM is defined as any light microscopy method that allows imaging at resolutions higher than the diffraction barrier. The diffraction barrier, or Abbe limit of detection, is expressed mathematically by κ = (2n sinα/λ), where λ is the wavelength of the emitted light, n is the refractive index of the medium containing the sample, and α is the angular aperture of the microscope.
When imaging a single fluorophore, which is less than a few nanometers in diameter, the resolving power limitation due to interactions between light and the medium can be defined as the point spread function. For visible light, this is roughly half the wavelength—approximately 250 nm for lateral resolution and roughly twice that in depth. This means that if two or more fluorophores are within a few hundred nanometers of one another, their images become blurred together, limiting resolution. In contrast, SRM approaches can achieve resolution from ~100 nm to <20 nm (Figure 1), nearing the resolution historically reserved for electron microscopy [1].
Four SRM technologies are now available: structured illumination microscopy (SIM), stimulated emission depletion (STED), stochastic optical reconstruction microscopy (STORM), and photoactivation localization microscopy (PALM) [1–3]. Each of these technologies benefits from the use of preferred fluorescent probes.
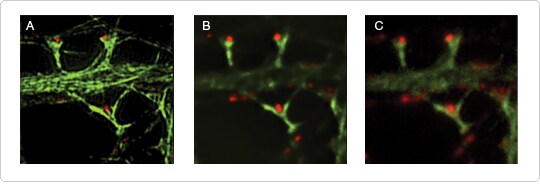
Figure 1. Comparison of super-resolution microscopy with wide-field deconvolution and standard wide-field microscopy. Alexa Fluor 488 dye–labeled microtubules and Alexa Fluor 568 dye–labeled synapsin in cultured hippocampal neurons (21DIV) were imaged with (A) the Nikon® N-SIM 3D structured illumination microscope; (B) wide-field deconvolution microscopy; or (C) standard wide-field microscopy. Images contributed by Gary Bassell, Emory University.
SIM: 3D and Live-cell Imaging
In SIM [4], structured light patterns are projected onto the sample to create Moiré fringes (interference patterns). Reconstruction software deciphers the images at about 2-fold higher resolution than the diffraction limit, or ~100 nm. Although the resolution of SIM alone is lower than that achieved by other SRM methods, it has several advantages. SIM can be used for imaging thicker sections, for 3D imaging, for live-cell imaging [5], and with fluorescent dyes, Qdot nanocrystals, and fluorescent proteins (Table 1).
Because SIM image resolution is impacted by probe quality, bright and photostable fluorophores such as Molecular Probes Alexa Fluor dyes and Qdot nanocrystals provide the highest-quality images. Fluorescent particles such as TetraSpeck™ microspheres (Figure 2) can be used for instrument calibration in the X, Y, and Z dimensions. Genetic tags for tubulin, actin, other structural proteins, and organelle markers provided by the CellLight reagents may be valuable for both live-cell and fixed-cell SIM applications and can provide both instrument calibration and experimental controls. SIM microscopes are available from Applied Precision (DeltaVision OMX® platform), Zeiss (ELYRA® S.1 and PS.1), and Nikon (N-SIM platform).
Table 1. Probes For Structured Illumination Microscopy.
| Product | Description |
|---|---|
| Alexa Fluor dyes and conjugates | Wide variety of labeling kits, conjugated primary and secondary antibodies, and other conjugates |
| Qdot nanocrystals | Wide variety of colors, labeling kits, conjugated secondary antibodies, and other conjugates |
| CellLight reagents | Genetically encoded cellular labeling reagents. GFP-, CFP-, and RFP-tagged versions of structural proteins, including tubulin, actin, histone 2B, and markers for a variety of different organelles. Ideal for instrument calibration, structural analysis, or experimental internal controls or positioning. Compatible with primary and stem cell labeling. |
| Probes for cytoskeletal proteins and Probes for organelles | Additional probes such as fluorescent phalloidin conjugates, TubulinTracker™ reagent, and MitoTracker dyes |
| TetraSpeck™ microspheres, 0.1 µm | Fluorescent blue, green, orange, or dark red. Ideal for calibration. |

Figure 2. Resolution limit improvement with structured illumination microscopy (SIM) versus traditional wide-field microscopy. Marked improvement in 2D (lateral) resolution is shown when using SIM (A) compared to wide-field microscopy (B), demonstrated with a group of four individual 100 nm Molecular Probes FluoSpheres microspheres (ex/em 505 nm/515 nm) on the Nikon® N-SIM platform using 488 nm excitation. The measured full-width half-maximum resolution for wide-field microscopy is ~250 nm, which improves to 110 nm with SIM. Not shown here, but similar gains are attained with axial or depth resolution—improving from 930 nm to 410 nm on the N-SIM platform.
Fluorophore Emission Isolation Approaches
STORM and PALM methods achieve high resolution by allowing for only a single fluorophore in a given region of interest to emit light at any given time [1]. In the case of STED, the excitation beam is constricted, limiting the excited volume to just a few fluors [1]. By eliminating emission overlap from nearby fluorophores, the centroid of the point spread function can be determined and used to locate the fluorophore within tens of nanometers. By using these approaches, the full image of the sample is obtained by layering multiple frames.
Two approaches to fluorophore emission isolation are possible. STED microscopy uses hardware; STORM AND PALM use a probe-based approach. See Table 2 for a range of reagents available for these technologies.
Table 2. Probes for STED Microscopy.
Probes for STORM, and dSTORM Microscopy.
STED: Imaging of Thick Sections
STED microscopy (Figure 3) uses two scanning laser beams. One beam provides the excitation light, while a second depletion beam de-excites the fluorophore to a nonfluorescent state, thereby shrinking the effective excited area and decreasing the apparent point spread function [6]. The depletion beam is aligned so it encircles the excitation beam like a donut and switches off any nearby fluorophores, and the full image is acquired by rastering across the sample. STED instrumentation gives X, Y resolutions of ~80 nm and a Z resolution comparable to that of confocal microscopy. Images can be acquired from samples 10–15 µm deep. The TCS STED CW microscope is available from Leica Microsystems.
For optimal CW-STED images, the fluorescent probe must match the excitation laser beam of the instrument (488 and 514 nm) and also respond to fluorescence depletion at the depletion beam wavelength (592 nm). We offerseveral compatible reagents, including the Oregon Green 488 and Alexa Fluor 488 and 594 dyes, and the fluorescent proteins YFP and Venus. The Alexa Fluor 594 dye has also been used to image brain tissue in a two-photon STED approach [7].
 | Figure 3. STED microscopy of intermediate filaments visualized with Oregon Green staining. PtK2 cells were methanol-fixed, and vimentin was labeled with Oregon Green 488 dye by indirect immunofluorescence. Confocal (A) and STED (B) images were acquired with a Leica TCS STED CW microscope. Images courtesy of Leica Microsystems. |
Single-molecule Detection by STORM and PALM
Whereas STED microscopy isolates fluorophore emission via hardware, STORM and PALM (Figures 4, 5) do so via photoswitching or photoactivating fluorescent probes, enabling single-molecule detection. In the STORM and PALM approaches, only a small subset of fluorophores is excited in the field at any given time. This is achieved by controlling the excitation light pulse and using a second wavelength to turn the probe off by photoswitching or photobleaching. This process is repeated thousands of times to compile the full image by stacking sequential frames.
STORM relies on photoswitching dyes [8,11], which consist of an emitter dye and an activator dye very close to each other. In excitation light, the emitter dye undergoes a reversible chemical reaction that interferes with fluorescence emission [12]. The reaction is reversed by energy transfer upon excitation of the activator dye, allowing photoswitching to occur repeatedly (200–300 times). Thus, by coordinating two different laser beams, the fluorophores emit light stochastically. Altering either the activator dye or the emission/reporter dye enables multiplexing.
Alexa Fluor 405 dye has been the workhorse activator dye for STORM. Good choices for emitter dyes include Alexa Fluor 647, 488, and 555, which match laser options for the Nikon N-STORM platform. Alternatively, single dyes, such as Alexa Fluor 647, have been used for photoswitching in a process called dSTORM [13]. Both STORM and dSTORM are dependent on thiol-containing media required for photoswitching.
In contrast to STORM, PALM relies on photoactivatable, photoconvertible, or photoswitchable fluorescent proteins to achieve probe blinking [9]. For example, 405 nm light has been used to activate GFP from a dark to a green species, to convert Eos from a green to a red species, or to photoswitch Dronpa from a dark to a fluorescent state [10]. A PALM instrument is available from Zeiss as the ELYRA® P.1 platform.
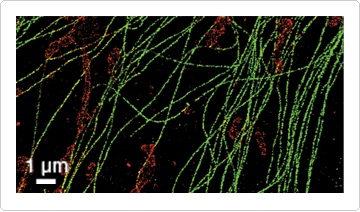
Figure 4. Two-color STORM image of microtubules (green) and mitochondria (red). BS-C-1 cells were aldehyde-fixed, reduced with sodium borohydride, and labeled with primary antibodies to tubulin and the mitochondrial marker Tom20. Secondary antibodies conjugated with the photoswitchable dye pairs Alexa Fluor 405/Alexa Fluor 647 and Cy®3/Alexa Fluor 647 were used to label tubulin and Tom20, respectively. Image courtesy of Xiaowei Zhuang, Harvard University.
 | Figure 5. dSTORM imaging of actin filaments. NIH-3T3 cells were stained with Alexa Fluor 647 phalloidin and imaged on the Zeiss ELYRA® P.1 system. (A) dSTORM image. (B) Wide-field image. Images contributed by Yilmaz Niyaz, CZ MicroImaging BioScience Scientific Support, Zeiss Labs, Munich. |
Advantages of Dyes vs. Fluorescent Proteins
STORM and PALM have advantages and disadvantages inherent to their affiliated probes. Because antibodies are still largely necessary as affinity reagents for dye labels, fluorescent proteins have an advantage in SRM applications because they are only ~2 nm from the protein target being imaged versus at least 7 nm for immunodetection, and generally do not have background staining. Though genetically encodable fluorescent proteins allow live-cell imaging and bypass the need for affinity reagents, they generally can only be photoswitched once or a few times, limiting attempts at image acquisition. However, this property can be beneficial for quantitative analyses.
Dyes are much brighter and more photostable than fluorescent proteins, but present hurdles for labeling structures in the absence of affinity reagents such as antibodies. While the ability to multiplex photoactivatable/photoswitchable fluorescent proteins remains limiting due their broad excitation/emission peaks, dyes are plentiful for multiplexing, even in SRM. Clearly, fluorescent proteins and fluorescent dyes will remain complementary tools for researchers performing SRM applications.
The Future of Fluorescence Microscopy
Although SRM technologies do not yet meet the resolution achieved by electron microscopy, improvements in cameras, optics, algorithms, and fluorescent probes are expected to bring the technology to even higher resolution. With further advances in live-cell imaging and multicolor analysis, SRM promises to add more clarity to our understanding of cellular processes. As researchers provide ever clearer images of cellular structures and dynamic events, diffraction-limited fluorescence microscopy will likely become regarded as vision impaired.
References
- Hell SW (2007) Science 316:1153–1158.
- Huang B, Bates M, Zhuang X (2009) Annu Rev Biochem 78:993–1016.
- Schermelleh L, Heintzmann R, Leonhardt H (2010) J Cell Biol 190:165–175.
- Gustafsson MG (2005) Proc Natl Acad Sci U S A 102:13081–13086.
- Kner P, Chhun BB, Griffis ER et al (2009) Nat Methods 6:339–342.
- Hell SW, Wichmann J (1994) Opt Lett 19:780–782.
- Ding J, Takasaki KT, Sabatini BL (2009) Neuron 63:429–437.
- Rust MJ, Bates M, Zhuang X (2006) Nat Methods 3:793–795.
- Betzig E, Patterson GH, Sougrat R et al (2006) Science 313:1642–1645.
- Day RN, Davidson MW (2009) Chem Soc Rev 38:2887–2921
- Bates M, Huang B, Dempsey GT et al (2007) Science 317:1749–1753.
- Dempsey GT, Bates M, Kowtoniuk WE et al (2009) J Am Chem Soc 131:18192–18193.
- Heilemann M, van de Linde S, Schuttpelz M et al (2008) Angew Chem Int Ed 47:6172–6176.
Learn more about:
- Super-Resolution Microscopy
- Alexa Fluor Dyes
- Probes for Cytoskeletal Proteins
- Probes for Organelles
- SIM Microscopy from Zeiss and Nikon
- STORM Microscopy from Nikon
- PALM Microscopy from Zeiss
Article download
Get a copy of this article as it appears in the print version of BioProbes 64.
For Research Use Only. Not for use in diagnostic procedures.