Search
Gene Expression Profiling and miRNA Discovery in a Carcinoma Model
With the recent realization that microRNAs (miRNAs), a class of small non-coding RNAs, are responsible for significant post-transcriptional regulation of gene expression, an additional area of inquiry has been added to comprehensive studies of gene expression. miRNAs negatively regulate expression by translational inhibition or cleavage of their targeted messenger RNAs (mRNAs) [1]. It is becoming clear that miRNAs are involved in regulating many diverse biological processes, and several recent publications have documented differential expression of miRNAs between malignant and normal tissues. miRNAs play a role in tumor formation, and their expression patterns may prove useful for tumor classification and treatment [2–3].
Here we describe a step-by-step strategy for parallel analysis of mRNA and miRNA expression using the Applied Biosystems and Ambion® tool set. A recent study about transcriptional and post-transcriptional gene regulation in a thyroid cancer model [4] illustrates the type of data that can be acquired from such studies. The researchers headed by Dr. Orla Sheils (Department of Histopathology, University of Dublin) looked at differences in gene expression between a normal thyroid cell line and two papillary thyroid carcinoma cell lines. Their investigation involved a step-by-step approach using microarray analysis, real-time PCR for both mRNA and miRNA targets, and bioinformatics tools to verify and correlate the results.
Here we describe a step-by-step strategy for parallel analysis of mRNA and miRNA expression using the Applied Biosystems and Ambion® tool set. A recent study about transcriptional and post-transcriptional gene regulation in a thyroid cancer model [4] illustrates the type of data that can be acquired from such studies. The researchers headed by Dr. Orla Sheils (Department of Histopathology, University of Dublin) looked at differences in gene expression between a normal thyroid cell line and two papillary thyroid carcinoma cell lines. Their investigation involved a step-by-step approach using microarray analysis, real-time PCR for both mRNA and miRNA targets, and bioinformatics tools to verify and correlate the results.
Papillary Thyroid Carcinoma Model
Papillary thyroid carcinoma (PTC) is the most common thyroid malignancy, accounting for 80–90% of cases [5]. Genetic studies suggest that the activation of chimeric RET oncogenes (ret/PTC) is one of the key first steps in thyroid cancer pathogenesis. At least 15 chimeric mRNAs involving ten distinct donor genes have been described. However, their exact role in the context of PTC remains unclear, and the complete repertoire of genes and signaling pathways involved in the pathogenesis of thyroid disease is still poorly defined. The authors of the study described here sought to identify the effect of the ret/PTC oncogene on transcription and post-transcriptional regulation of gene expression in tumorigenesis.
Isolation of Total RNA Including the MicroRNA Fraction
Many commonly used RNA isolation methods were developed before there was much interest in small RNAs, and they do not quantitatively recover the small RNA fraction. In this study, the
Ambion mirVana™ miRNA Isolation Kit was used to isolate total RNA, including the miRNA fraction, from the following thyroid-derived cell lines:
- Nthy-ori 3-1: normal thyroid follicular epithelial cell line
- Nthy-ret: Nthy-ori 3-1 cells transfected with ret/PTC-1 using a mammalian expression vector system
- TPC-1: cell line derived from a PTC that expresses the ret/PTC-1 oncogene [H4-(CCDC6)-RET].
Expression Profiling of mRNA and miRNA
The most common techniques for mRNA expression profiling rely on microarrays, which consist of large numbers of immobilized nucleic acids specific for individual sequences in labeled samples derived from RNA. This is a prudent approach for profiling the tens of thousands of individual mRNAs that are expressed in any given sample. Microarrays also can be used to evaluate miRNA expression levels, but because miRNAs are numbered in the hundreds, instead of thousands, real-time PCR represents a simpler, more direct alternative. Because of their small size, however, real-time PCR of miRNA requires innovative primer/probe design to obtain meaningful results.
mRNA Expression Profiling using the Applied Biosystems Expression Array System
In this study, mRNA expression analysis was performed using the Applied Biosystems Expression Array System (see sidebar), which consists of chemiluminescent sample labeling and detection kits and the 1700 Chemiluminescent Microarray Analyzer. Hybridization signals were quantified and corrected for background using the software resident on the 1700 instrument. Statistical analysis of the data revealed that in ret/PTC cell lines compared to the normal thyroid cell line, 47 genes were upregulated, and 29 genes were downregulated.
Functional Interpretation of the mRNA Expression Data
The differentially expressed mRNAs were then categorized according to their molecular function using the free online database known as PANTHER™ (Protein ANalysis THrough Evolutionary Relationships) Classification System (Figure 1). Among the upregulated group, genes involved in biological processes, such as cell differentiation and proliferation, were highly represented—including genes that have been shown to play critical roles in tumorigenesis or cell survival.
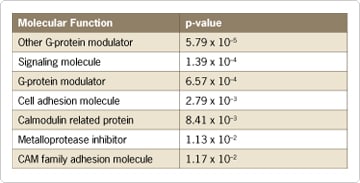
Figure 1. Significance of Molecular Functions of Differentially Expressed Genes in ret/PTC Cells versus Normal Thyroid Cell Lines. The Chemiluminescent RT-IVT Kit v2.0 (now replaced with the Applied Biosystems NanoAmp™ RT-IVT Labeling Kit) was used to generate digoxigenin-labeled amplified RNA from triplicate samples of total RNA (2 µg) from the normal thyroid cell line and cell lines with the ret/PTC oncogene. Labeled RNA samples were hybridized to Applied Biosystems Human Genome Survey Microarray V2.0, and the Chemiluminescent Labeling Kit was used to detect the digoxigenin-labeled sample. Signal was collected from the microarrays using the Applied Biosystems 1700 Chemiluminescent Microarray Analyzer. Statistical analysis and normalization were performed using R version 1.9.1. p-values were determined using an ANOVA test and were adjusted for multiple comparisons with a cut-off False Discovery Rate (FDR) <0.1. p-values <0.01 and fold change >2 were considered statistically significant. The differentially expressed genes were categorized according to their molecular function using PANTHER™ Classification System, and the distribution of the fold change in expression values within each functional group was compared with a theoretically random distribution and statistically evaluated using the Mann-Whitney U-test [6].
A high proportion of the genes that were found to be downregulated in the thyroid carcinoma cell model were involved in cell structure and motility. Of particular interest was the discovery that DROSHA was downregulated in the PTC cell lines. This differential expression may affect the production of miRNA and consequently play a role in the regulation of other genes.
Profiling MicroRNA Expression
miRNA expression was evaluated using Applied Biosystems reverse transcription reagents and a collection of 157 TaqMan® MicroRNA Assays, which targeted the majority of publically available human miRNAs. PCR amplification, and signal detection was performed on the Applied Biosystems 7900HT Fast Real-Time PCR System. When compared to normal thyroid cells, 21 miRNAs were found to be overexpressed and 14 miRNAs underexpressed in cell lines with the ret/PTC rearrangement (Figure 2). Furthermore, a unique miRNA expression signature was identified that differentiates the normal cell line and the PTC model system.
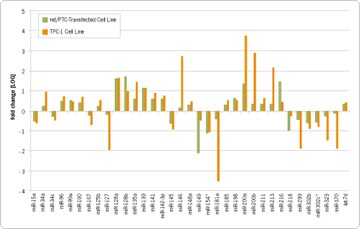
Figure 2. Differential Expression of miRNAs in a Normal Thyroid Cell Line Compared to Cell Lines with the ret/PTC Rearrangement. Fold change of miRNA expression in a ret/PTC-transfected cell line (green) and TPC-1 cell line (orange) compared to a normal thyroid cell line using the DDCT method. Data were normalized to hsa-let-7a as the endogenous control and to the normal thyroid cell line (Nthy-ori) as a normal control (calibrator). The log of the relative quantitation (RQ) values was used to plot the relative fold change.
Validation of mRNA Microarray Results and Targeted miRNA Analysis
Regardless of the method used for expression profiling, further experimentation is typically indicated—either to validate differential expression with an independent technique in the case of mRNA microarray analysis, or to conduct targeted miRNA profiling of miRNAs identified in an initial miRNA screen. For this study, TaqMan® Gene Expression Assays were used to validate mRNA expression profiling results, and the expression of individual miRNAs was analyzed using TaqMan MicroRNA Assays (data not shown).
The next step was to compare the list of differentially expressed genes identified in the mRNA microarray screen with a list of putative miRNA targets. Using databases for miRNA target prediction (i.e., miRBase, PICTAR, and TARGETSCAN), the authors of this study were able to generate a list of 15 potential mRNA binding partners for several of the miRNAs that were observed to be differentially expressed in cell lines with the ret/PTC rearrangement (Figure 3). Notably these miRNAs potentially regulate a number of genes that govern thyroid function, cell signaling, and cell cycle control, and have implications in thyroid cancer. The significance of such a correlation between the data sets is purely speculative at this point but provides an interesting avenue for further investigation.
Figure 3. Differentially Expressed miRNAs and Their Potential mRNA Targets. These miRNAs were differentially expressed in ret/PTC cells compared to normal thyroid cell lines. The list of potential targets was identified using miRBase, PICTAR, and TARGETSCAN databases for miRNA target prediction.
mRNA Expression Profiling using the Applied Biosystems Expression Array System
In this study, mRNA expression analysis was performed using the Applied Biosystems Expression Array System (see sidebar), which consists of chemiluminescent sample labeling and detection kits and the 1700 Chemiluminescent Microarray Analyzer. Hybridization signals were quantified and corrected for background using the software resident on the 1700 instrument. Statistical analysis of the data revealed that in ret/PTC cell lines compared to the normal thyroid cell line, 47 genes were upregulated, and 29 genes were downregulated.
Functional Interpretation of the mRNA Expression Data
The differentially expressed mRNAs were then categorized according to their molecular function using the free online database known as PANTHER™ (Protein ANalysis THrough Evolutionary Relationships) Classification System (Figure 1). Among the upregulated group, genes involved in biological processes, such as cell differentiation and proliferation, were highly represented—including genes that have been shown to play critical roles in tumorigenesis or cell survival.
Figure 1. Significance of Molecular Functions of Differentially Expressed Genes in ret/PTC Cells versus Normal Thyroid Cell Lines. The Chemiluminescent RT-IVT Kit v2.0 (now replaced with the Applied Biosystems NanoAmp™ RT-IVT Labeling Kit) was used to generate digoxigenin-labeled amplified RNA from triplicate samples of total RNA (2 µg) from the normal thyroid cell line and cell lines with the ret/PTC oncogene. Labeled RNA samples were hybridized to Applied Biosystems Human Genome Survey Microarray V2.0, and the Chemiluminescent Labeling Kit was used to detect the digoxigenin-labeled sample. Signal was collected from the microarrays using the Applied Biosystems 1700 Chemiluminescent Microarray Analyzer. Statistical analysis and normalization were performed using R version 1.9.1. p-values were determined using an ANOVA test and were adjusted for multiple comparisons with a cut-off False Discovery Rate (FDR) <0.1. p-values <0.01 and fold change >2 were considered statistically significant. The differentially expressed genes were categorized according to their molecular function using PANTHER™ Classification System, and the distribution of the fold change in expression values within each functional group was compared with a theoretically random distribution and statistically evaluated using the Mann-Whitney U-test [6].
A high proportion of the genes that were found to be downregulated in the thyroid carcinoma cell model were involved in cell structure and motility. Of particular interest was the discovery that DROSHA was downregulated in the PTC cell lines. This differential expression may affect the production of miRNA and consequently play a role in the regulation of other genes.
Profiling MicroRNA Expression
miRNA expression was evaluated using Applied Biosystems reverse transcription reagents and a collection of 157 TaqMan® MicroRNA Assays, which targeted the majority of publically available human miRNAs. PCR amplification, and signal detection was performed on the Applied Biosystems 7900HT Fast Real-Time PCR System. When compared to normal thyroid cells, 21 miRNAs were found to be overexpressed and 14 miRNAs underexpressed in cell lines with the ret/PTC rearrangement (Figure 2). Furthermore, a unique miRNA expression signature was identified that differentiates the normal cell line and the PTC model system.
Figure 2. Differential Expression of miRNAs in a Normal Thyroid Cell Line Compared to Cell Lines with the ret/PTC Rearrangement. Fold change of miRNA expression in a ret/PTC-transfected cell line (green) and TPC-1 cell line (orange) compared to a normal thyroid cell line using the DDCT method. Data were normalized to hsa-let-7a as the endogenous control and to the normal thyroid cell line (Nthy-ori) as a normal control (calibrator). The log of the relative quantitation (RQ) values was used to plot the relative fold change.
Validation of mRNA Microarray Results and Targeted miRNA Analysis
Regardless of the method used for expression profiling, further experimentation is typically indicated—either to validate differential expression with an independent technique in the case of mRNA microarray analysis, or to conduct targeted miRNA profiling of miRNAs identified in an initial miRNA screen. For this study, TaqMan® Gene Expression Assays were used to validate mRNA expression profiling results, and the expression of individual miRNAs was analyzed using TaqMan MicroRNA Assays (data not shown).
The next step was to compare the list of differentially expressed genes identified in the mRNA microarray screen with a list of putative miRNA targets. Using databases for miRNA target prediction (i.e., miRBase, PICTAR, and TARGETSCAN), the authors of this study were able to generate a list of 15 potential mRNA binding partners for several of the miRNAs that were observed to be differentially expressed in cell lines with the ret/PTC rearrangement (Figure 3). Notably these miRNAs potentially regulate a number of genes that govern thyroid function, cell signaling, and cell cycle control, and have implications in thyroid cancer. The significance of such a correlation between the data sets is purely speculative at this point but provides an interesting avenue for further investigation.
Figure 3. Differentially Expressed miRNAs and Their Potential mRNA Targets. These miRNAs were differentially expressed in ret/PTC cells compared to normal thyroid cell lines. The list of potential targets was identified using miRBase, PICTAR, and TARGETSCAN databases for miRNA target prediction.
The Next Step: Functional Analysis
Plans are underway to further investigate the identified miRNA targets in the PTC-model using a novel algorithm for animal miRNA target prediction. The function of the miRNAs will be examined using synthetic miRNA mimics (
Ambion Pre-miR™ miRNA Precursors) and specific miRNA inhibitors (
Ambion Anti-miR™ miRNA Inhibitors) to upregulate and downregulate the expression of particular miRNAs while monitoring for commensurate mRNA and protein expression changes in the candidate target (see sidebar, Assess Biological Effects of miRNAs).
A Complete Solution for mRNA and miRNA Expression Studies
Applied Biosystems offers a complete suite of tools for the analysis of gene expression and regulation including sample preparation, mRNA and miRNA expression analysis, and detailed data analysis.
Scientific Contributors
Astrid Ferlinz, Simone Günther, Alexander Sartori, Jon Sherlock • Applied Biosystems
We greatly appreciate the opportunity to collaborate with Drs. Orla Sheils and John O’Leary from the Department of Histopathology of the University of Dublin, Ireland, and the following scientists in their groups, Susanne Cahill, Paul Smyth, Richard Flavin, Sinead Aherne, Karen Denning, Stephen Finn, Esther O’Regan, and Jinghuan Li.
Scientific Contributors
Astrid Ferlinz, Simone Günther, Alexander Sartori, Jon Sherlock • Applied Biosystems
We greatly appreciate the opportunity to collaborate with Drs. Orla Sheils and John O’Leary from the Department of Histopathology of the University of Dublin, Ireland, and the following scientists in their groups, Susanne Cahill, Paul Smyth, Richard Flavin, Sinead Aherne, Karen Denning, Stephen Finn, Esther O’Regan, and Jinghuan Li.
Isolate Total RNA Including miRNAThe
mirVana™ miRNA Isolation Kit was developed specifically for the purification of total RNA (size range: kilobases down to 10mers). The kit also provides reagents and a procedure to isolate an enriched population of RNA that are 200 bases and smaller, which enhances the sensitivity of small RNA detection by solution hybridization, Northern analysis, and other methods. This glass fiber filter-based kit is compatible with virtually all cell and tissue types and miRNA applications.