Search
Page Contents
The fundamental role of cell–cell and cell–matrix adhesion in the morphology and development of organisms, organs and tissues has made identification of molecular mediators of cell adhesion an important research focus in cell biology and immunology.![]() The useful review by Löster and Hortstkorte describes a number of different assays that can detect cell adhesion.
The useful review by Löster and Hortstkorte describes a number of different assays that can detect cell adhesion.![]() In a typical fluorescence-based cell adhesion assay, unlabeled cell monolayers in multiwell plates are incubated with fluorescently labeled cells and then washed to separate the adherent and nonadherent populations. Cell adhesion can then be determined simply by correlating the retained fluorescence with cell number. An ideal fluorescent marker will retain proportionality between fluorescence and cell number and introduce minimal interference with the cell adhesion process. Because adhesion is a cell-surface phenomenon, cytoplasmic markers that can be passively loaded are preferable to compounds that label cell-surface molecules, provided they are retained in the cell for the duration of the experiment or their leakage rate can be independently measured. Adhesion of fluorescent dye–labeled cells to matrices such as bone
In a typical fluorescence-based cell adhesion assay, unlabeled cell monolayers in multiwell plates are incubated with fluorescently labeled cells and then washed to separate the adherent and nonadherent populations. Cell adhesion can then be determined simply by correlating the retained fluorescence with cell number. An ideal fluorescent marker will retain proportionality between fluorescence and cell number and introduce minimal interference with the cell adhesion process. Because adhesion is a cell-surface phenomenon, cytoplasmic markers that can be passively loaded are preferable to compounds that label cell-surface molecules, provided they are retained in the cell for the duration of the experiment or their leakage rate can be independently measured. Adhesion of fluorescent dye–labeled cells to matrices such as bone ![]() can be directly observed by fluorescence microscopy using cells loaded with the permeant live-cell tracers described in Membrane-Permeant Reactive Tracers—Section 14.2 and Viability and Cytotoxicity Assay Reagents—Section 15.2 or the lipophilic dyes in Tracers for Membrane Labeling—Section 14.4. Alternatively, high molecular weight, cell-impermeant, fluorescent dextrans (Fluorescent and Biotinylated Dextrans—Section 14.5) have been used to define the area outside of adherent cells, with the adherent cells themselves remaining unstained.
can be directly observed by fluorescence microscopy using cells loaded with the permeant live-cell tracers described in Membrane-Permeant Reactive Tracers—Section 14.2 and Viability and Cytotoxicity Assay Reagents—Section 15.2 or the lipophilic dyes in Tracers for Membrane Labeling—Section 14.4. Alternatively, high molecular weight, cell-impermeant, fluorescent dextrans (Fluorescent and Biotinylated Dextrans—Section 14.5) have been used to define the area outside of adherent cells, with the adherent cells themselves remaining unstained.![]() This same "negative-staining" method can also be used to assess cell spreading and progress toward confluency.
This same "negative-staining" method can also be used to assess cell spreading and progress toward confluency.
Cell Adhesion Assays Using Enzyme Substrates
Essentially all of the esterase substrates in Esterase substrates for cell viability studies—Table 15.1 useful for monitoring cell viability can also be used for studying cell adhesion. As with cell viability studies, calcein AM (C1430, C3099, C3100MP) appears to best satisfy the criteria for assaying cell adhesion ![]() and to have the least effect on cell viability and other cell functions.
and to have the least effect on cell viability and other cell functions.![]() The results obtained with leukocyte adhesion assays using calcein AM correlate well with those obtained with 51Cr assays,
The results obtained with leukocyte adhesion assays using calcein AM correlate well with those obtained with 51Cr assays,![]() but the calcein AM protocols take less time and avoid the special handling required when using radioactive material. Calcein AM has been used in numerous cell adhesion assays, including those designed to measure:
but the calcein AM protocols take less time and avoid the special handling required when using radioactive material. Calcein AM has been used in numerous cell adhesion assays, including those designed to measure:
- Binding of labeled Jurkat cells to vascular cell adhesion molecule-1 (VCAM1) in cell membrane preparations
- Effects of E-selectin–binding peptides
and integrins
- Integrin-mediated cell adhesion in transfected K562 cells
- Leukocyte
- Monocyte adhesion in HIV-infected cells
Other fluorogenic esterase substrates that have been used to assess cell adhesion include BCECF AM ![]() (B1150, B1170, B3051; BCECF), carboxyfluorescein diacetate
(B1150, B1170, B3051; BCECF), carboxyfluorescein diacetate ![]() (5(6)-CFDA, C195), fluorescein diacetate
(5(6)-CFDA, C195), fluorescein diacetate ![]() (F1303), the succinimidyl ester of carboxyfluorescein diacetate
(F1303), the succinimidyl ester of carboxyfluorescein diacetate ![]() (5(6)-CFDA, SE; CFSE; C1157) and CellTracker Green CMFDA
(5(6)-CFDA, SE; CFSE; C1157) and CellTracker Green CMFDA ![]() (C2925, C7025), all of which are discussed in Viability and Cytotoxicity Assay Reagents—Section 15.2. Because of the possibility of slow calcein leakage from calcein AM–labeled cells, CellTracker Green CMFDA and 5(6)-CFDA SE are recommended for quantitative adhesion or aggregation assays that require incubation for more than about an hour.
(C2925, C7025), all of which are discussed in Viability and Cytotoxicity Assay Reagents—Section 15.2. Because of the possibility of slow calcein leakage from calcein AM–labeled cells, CellTracker Green CMFDA and 5(6)-CFDA SE are recommended for quantitative adhesion or aggregation assays that require incubation for more than about an hour.![]() Use of a combination of a green-fluorescent dye (usually calcein AM, BCECF AM or CMFDA) and a red-fluorescent dye—especially chloromethyl SNARF-1 acetate (C6826), SNARF-1 carboxylic acid, acetate, succinimidyl ester (S22801, Assays for Cell Enumeration, Cell Proliferation and Cell Cycle—Section 15.4) or carboxynaphthofluorescein diacetate (C13196, Viability and Cytotoxicity Assay Reagents—Section 15.2)—enables two-color measurements of adhesion or cell–cell aggregation in mixed cell cultures.
Use of a combination of a green-fluorescent dye (usually calcein AM, BCECF AM or CMFDA) and a red-fluorescent dye—especially chloromethyl SNARF-1 acetate (C6826), SNARF-1 carboxylic acid, acetate, succinimidyl ester (S22801, Assays for Cell Enumeration, Cell Proliferation and Cell Cycle—Section 15.4) or carboxynaphthofluorescein diacetate (C13196, Viability and Cytotoxicity Assay Reagents—Section 15.2)—enables two-color measurements of adhesion or cell–cell aggregation in mixed cell cultures.![]() Calcein AM has also been used in a quantitative microplate assay of spreading of adherent cells on artificial or biological surfaces.
Calcein AM has also been used in a quantitative microplate assay of spreading of adherent cells on artificial or biological surfaces.![]()
In their review,![]() Löster and Hortstkorte describe the use of a number of other fluorogenic substrates for quantitating cell adhesion; among these are fluorescein diphosphate (F2999, Detecting Enzymes That Metabolize Phosphates and Polyphosphates—Section 10.3) and ELF 97 phosphate (Phosphatase-Based Signal Amplification Techniques—Section 6.3) for lysosomal and membrane phosphatases, as well as various substrates for glycosidases (Detecting Glycosidases—Section 10.2) and oxidases (Substrates for Oxidases, Including Amplex Red Kits—Section 10.5).
Löster and Hortstkorte describe the use of a number of other fluorogenic substrates for quantitating cell adhesion; among these are fluorescein diphosphate (F2999, Detecting Enzymes That Metabolize Phosphates and Polyphosphates—Section 10.3) and ELF 97 phosphate (Phosphatase-Based Signal Amplification Techniques—Section 6.3) for lysosomal and membrane phosphatases, as well as various substrates for glycosidases (Detecting Glycosidases—Section 10.2) and oxidases (Substrates for Oxidases, Including Amplex Red Kits—Section 10.5).
Vybrant Cell Adhesion Assay Kit
The Vybrant Cell Adhesion Assay Kit (V13181) utilizes calcein AM to provide a fast and sensitive method for measuring cell–cell or cell–substratum adhesion.![]() Calcein AM is nonfluorescent but, once loaded into cells, is cleaved by endogenous esterases to produce calcein, a highly fluorescent and well-retained dye. Calcein provides a bright green-fluorescent, pH-independent, cytoplasmic cell marker that does not appear to affect the cell adhesion process.
Calcein AM is nonfluorescent but, once loaded into cells, is cleaved by endogenous esterases to produce calcein, a highly fluorescent and well-retained dye. Calcein provides a bright green-fluorescent, pH-independent, cytoplasmic cell marker that does not appear to affect the cell adhesion process.![]()
To perform this assay, samples of calcein AM–labeled cells are added to monolayers of unlabeled cells in a microplate. Following incubation to allow the labeled cells to adhere to the unlabeled cells, the samples are washed to remove any nonadhering labeled cells (Figure 15.6.1). The calcein fluorescence in this sample, as compared with that of an unlabeled control sample, can then be used to calculate the number of adherent cells. The absorption/emission maxima of calcein (~494/517 nm) are ideally suited for detection by a fluorescence microplate reader equipped with standard fluorescein optical filters.
In addition to calcein AM, the Vybrant Cell Adhesion Assay Kit includes SYTOX Green nucleic acid stain, an easy-to-use dead-cell indicator for assessing overall health of cells prior to performing the cell adhesion assay. As a fluorescent substitute for trypan blue, this high-affinity nucleic acid stain easily penetrates cells that have compromised membranes but will not cross the membranes of live cells. Upon binding to nucleic acids, the SYTOX Green dye exhibits a >500-fold fluorescence enhancement and can be observed with standard fluorescein optical filters (excitation/emission ~504/523 nm).
The Vybrant Cell Adhesion Assay Kit contains sufficient reagents to perform about 1000 assays using a fluorescence microplate reader, including:
- Calcein AM
- SYTOX Green nucleic acid stain, an easy-to-use green-fluorescent indicator of the overall health of cells prior to performing the cell adhesion assay
- Detailed protocols (Vybrant Cell Adhesion Assay Kit)
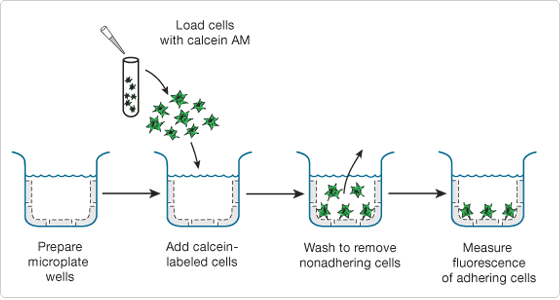
Figure 15.6.1 Principle of the Vybrant Cell Adhesion Assay Kit (V13181). Microplate wells may be left untreated or precoated with extracellular matrix proteins, antibodies or other reagents.
Cell Adhesion Assays Using the CyQUANT Cell Proliferation Assay Kits
Our CyQUANT Cell Proliferation Assay Kits (C7026, C35006, C35007, C35011, C35012; Assays for Cell Enumeration, Cell Proliferation and Cell Cycle—Section 15.4) can also serve as important tools for quantitating both cell–cell and cell–surface adhesion. The CyQUANT GR, CyQUANT NF and CyQUANT Direct assay detect total nucleic acids in cells, with a linear detection range from 50 to 50,000 cells per well and from 100 to 20,000 cells per well, respectively (Figure 15.6.2). To quantitate cell–surface adhesion, cells are simply permitted to adhere to the surface, gently washed to remove nonadherent cells and then analyzed to determine total nucleic acids in the adherent cells using the CyQUANT assay protocol. As a control, the fluorescence of the total number of cells added to the well before the wash step can be determined by the same assay method to yield the percentage of adherent cells. It should also be possible to extend the CyQUANT assay to studies of cell–cell adhesion by quantitating both the number of surface-adhering cells originally plated and the number of total cells after a second cell line has been introduced and allowed to adhere. A similar assay for cell adhesion based on DAPI (D1306, D3571, D21490; Nucleic Acid Stains—Section 8.1) has been reported.![]()
 | Figure 15.6.2 Quantitation of NIH 3T3 fibroblasts using the CyQUANT Cell Proliferation Assay Kit (C7026). Fluorescence measurements were made using a microplate reader with excitation at 485 nm and emission detection at 530 nm. The linear range of the assay under these conditions is from 50 to 50,000 cells per 200 µL sample. The inset shows the linearity that can be obtained at very low numbers of cells. |
Microsphere Adhesion Assays
In a novel method for studying adhesion in live neural tissue slices, including hippocampal slices, fluorescent 4 µm microspheres (F8858, F8859; Microspheres—Section 6.5) are coated with isolated cell membranes from dissociated live cells. The membrane-coated microspheres are then seeded on live tissue slices, and after a short incubation time, the nonadherent microspheres are eliminated by washing (Figure 15.6.3). The pattern of tissue labeling of adherent microspheres can then be visualized by epifluorescence microscopy.![]() A flow cytometry–based microsphere adhesion assay has also been developed to determine integrin-specific adhesion, as well as to analyze integrin-mediated adhesion defects in B-lineage acute lymphoblastic leukemia.
A flow cytometry–based microsphere adhesion assay has also been developed to determine integrin-specific adhesion, as well as to analyze integrin-mediated adhesion defects in B-lineage acute lymphoblastic leukemia.![]() FluoSpheres microspheres of different colors (or sizes or intensities) (Microspheres—Section 6.5) can be used for simultaneous labeling of different adhesion factors.
FluoSpheres microspheres of different colors (or sizes or intensities) (Microspheres—Section 6.5) can be used for simultaneous labeling of different adhesion factors.
 | Figure 15.6.3 Schematic view of the microsphere adhesion assay. Membrane-coated microspheres are added to a living tissue slice in a drop of medium (panel A). The microspheres disperse through the medium and eventually cover the tissue slice (panels B and C). After incubation, nonadherent microspheres are removed by extensive washing, and the sample is ready for analysis by fluorescence microscopy (panel D). |
Fluorescent Fibrinogen
Fibrinogen is a key component in the blood clotting process and can support both platelet–platelet and platelet–surface interactions by binding to the glycoprotein IIb-IIIa (GPIIb-IIIa) receptor ![]() (also called integrin αIIbβ3). Although the mechanism is not well understood, activation of GPIIb-IIIa is required for fibrinogen binding, which leads to platelet activation, adhesion, spreading and microfilament reorganization of human endothelial cells in vitro.
(also called integrin αIIbβ3). Although the mechanism is not well understood, activation of GPIIb-IIIa is required for fibrinogen binding, which leads to platelet activation, adhesion, spreading and microfilament reorganization of human endothelial cells in vitro.![]() Soluble fibrinogen binds to its receptor with a Ca2+-dependent apparent Kd of 0.18 µM.
Soluble fibrinogen binds to its receptor with a Ca2+-dependent apparent Kd of 0.18 µM.![]() This binding is apparently mediated by the tripeptide sequence Arg–Gly–Asp (RGD), found both in fibrinogen and fibronectin, as well as some other proteins.
This binding is apparently mediated by the tripeptide sequence Arg–Gly–Asp (RGD), found both in fibrinogen and fibronectin, as well as some other proteins.![]()
Fluorescently labeled fibrinogen has proven to be a valuable tool for investigating platelet activation and subsequent fibrinogen binding. Fluorescein fibrinogen has been used to identify activated platelets by flow cytometry.![]() The binding of fluorescein fibrinogen to activated platelets has been shown to be saturable and can be inhibited completely by underivatized fibrinogen.
The binding of fluorescein fibrinogen to activated platelets has been shown to be saturable and can be inhibited completely by underivatized fibrinogen.![]() The preferential binding and accumulation of fluorescein fibrinogen at the endothelial border of venular blood vessels has been studied by quantitative fluorescence microscopy.
The preferential binding and accumulation of fluorescein fibrinogen at the endothelial border of venular blood vessels has been studied by quantitative fluorescence microscopy.![]() We offer five fluorescent conjugates of human fibrinogen, which are useful for investigating platelet activation and subsequent fibrinogen binding using fluorescence microscopy or flow cytometry (Figure 15.6.4):
We offer five fluorescent conjugates of human fibrinogen, which are useful for investigating platelet activation and subsequent fibrinogen binding using fluorescence microscopy or flow cytometry (Figure 15.6.4):
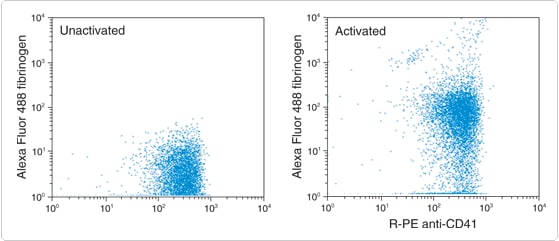
Figure 15.6.4 Interaction of fluorescently labeled fibrinogen with activated platelets. Whole blood was first incubated with an R-phycoerythrin (R-PE)–labeled anti-CD41 antibody to label the platelets. 20 µM adenosine 5'-diphosphate (ADP) was added in order to activate the platelets, then 2 µg/mL Alexa Fluor 488 fibrinogen (F13191) was added and incubated with the sample for 5 minutes. Cells were analyzed by flow cytometry using excitation at 488 nm. Both activated and unactivated platelets show binding of the anti-CD41 antibody; however, only the activated platelets show strong binding by fibrinogen. A total of 5000 platelets are shown in each experiment.
Fluorescent Gelatin and Collagen
Collagen is a major component of the extracellular matrix and, in vertebrates, constitutes approximately 25% of total cellular protein. This important protein not only serves a structural role, but also is important in cell adhesion and migration. Specific collagen receptors, fibronectin and a number of other proteins involved in cell–cell and cell–surface adhesion have been demonstrated to bind collagen and gelatin ![]() (denatured collagen). Fluorescent conjugates of gelatin and collagen for use in studying collagen-binding proteins and collagen metabolism, as well as gelatinases and collagenases (Detecting Peptidases and Proteases—Section 10.4), which are metalloproteins that digest gelatin and collagen.
(denatured collagen). Fluorescent conjugates of gelatin and collagen for use in studying collagen-binding proteins and collagen metabolism, as well as gelatinases and collagenases (Detecting Peptidases and Proteases—Section 10.4), which are metalloproteins that digest gelatin and collagen.
We offer two green-fluorescent gelatin conjugates—fluorescein gelatin (G13187) and Oregon Green 488 gelatin (G13186). When compared with the fluorescein conjugate, the Oregon Green 488 conjugate exhibits almost identical fluorescence spectra but its fluorescence is much more photostable and less pH dependent. We also offer collagen-coated FluoSpheres yellow-green–fluorescent microspheres (F20892, F20893; Probes for Following Receptor Binding and Phagocytosis—Section 16.1). By analogy to results obtained with fluorescein conjugates of these proteins, these highly fluorescent gelatin conjugates and collagen-coated microspheres are potentially useful for:
- Following integrin-mediated phagocytosis
- Localizing surface fibronectin on cultured cells
- Studying fibronectin–gelatin interactions in solution using fluorescence polarization
- Visualizing gelatinase activity using in situ gel zymography
Direct Detection of Chemotaxis Using Cell Counting
Chemotaxis, defined as directed cell motion toward an extracellular gradient, plays an important role during fertilization, inflammation, wound healing and hematopoiesis.![]() Chemotaxis is typically assayed by determining the number of viable cells that have migrated through a special "chemotaxis chamber." A 96-well disposable chemotaxis chamber is reported to be suitable for fluorescence-based assays that are faster, less labor intensive and more sensitive than visually detected migration assays.
Chemotaxis is typically assayed by determining the number of viable cells that have migrated through a special "chemotaxis chamber." A 96-well disposable chemotaxis chamber is reported to be suitable for fluorescence-based assays that are faster, less labor intensive and more sensitive than visually detected migration assays.![]() The probes used to follow chemotaxis in live cells are often the same esterase substrates that are used for assaying cell viability and cell adhesion (Esterase substrates for cell viability studies—Table 15.1), including calcein AM
The probes used to follow chemotaxis in live cells are often the same esterase substrates that are used for assaying cell viability and cell adhesion (Esterase substrates for cell viability studies—Table 15.1), including calcein AM ![]() (C1430, C3099, C3100MP), BCECF AM
(C1430, C3099, C3100MP), BCECF AM ![]() (B1150, B1170, B3051) and CellTracker Green CMFDA
(B1150, B1170, B3051) and CellTracker Green CMFDA ![]() (C2925, C7025). Calcein AM does not interfere with lymphocyte proliferation or with granulocyte or neutrophil chemotaxis or superoxide production,
(C2925, C7025). Calcein AM does not interfere with lymphocyte proliferation or with granulocyte or neutrophil chemotaxis or superoxide production,![]() and, unlike BCECF AM, calcein AM does not affect chemotaxis in leukocytes.
and, unlike BCECF AM, calcein AM does not affect chemotaxis in leukocytes.![]()
Because chemotaxis involves translocation of whole cells, assays that simply count cell numbers—such as our CyQUANT Cell Proliferation Assay Kit (C7026, Assays for Cell Enumeration, Cell Proliferation and Cell Cycle—Section 15.4)—are also quite reliable for following chemotaxis.![]() In addition, the green-fluorescent SYTO 13 dye (S7575, Nucleic Acid Stains—Section 8.1) has been used to track the co-migration of separately stained populations of neutrophils using opposing gradients of leukotriene B4 and interleukin 8 as the chemoattractants.
In addition, the green-fluorescent SYTO 13 dye (S7575, Nucleic Acid Stains—Section 8.1) has been used to track the co-migration of separately stained populations of neutrophils using opposing gradients of leukotriene B4 and interleukin 8 as the chemoattractants.![]()
Probes for Chemotaxis Receptors
We prepare the fluorescein conjugate of the chemotactic hexapeptide N-formyl-Nle-Leu-Phe-Nle-Tyr-Lys (F1314), which binds to the fMLF receptor ![]() (Probes for Following Receptor Binding and Phagocytosis—Section 16.1). We also offer fluorescein-labeled casein (C2990), which has been used to demonstrate casein-specific chemotaxis receptors in human neutrophils and monocytes with flow cytometry.
(Probes for Following Receptor Binding and Phagocytosis—Section 16.1). We also offer fluorescein-labeled casein (C2990), which has been used to demonstrate casein-specific chemotaxis receptors in human neutrophils and monocytes with flow cytometry.![]() Neutrophils activated with phorbol myristate acetate have been shown to undergo a dose-dependent increase in binding of fluorescein-labeled casein.
Neutrophils activated with phorbol myristate acetate have been shown to undergo a dose-dependent increase in binding of fluorescein-labeled casein.![]()
Multidrug resistance (MDR) is a phenomenon representing a complex group of biological processes that are of growing interest in both clinical and experimental oncology.![]() The MDR phenotype is characterized by the acquired resistance of tumor cells to structurally and functionally dissimilar anticancer drugs. Among the many mechanisms contributing to this multidrug resistance are the following:
The MDR phenotype is characterized by the acquired resistance of tumor cells to structurally and functionally dissimilar anticancer drugs. Among the many mechanisms contributing to this multidrug resistance are the following:
- Amplification of genes encoding drug-metabolizing enzymes
- Elevated levels of glutathione and glutathione-conjugating enzymes
- Mutated DNA topoisomerases
- Overexpression of plasma membrane ATP-dependent drug efflux pumps
Based on substrate and inhibitor profiles, at least four different plasma membrane ATP-dependent drug efflux pumps have been identified.![]() The activity of the verapamil-sensitive P-glycoprotein (Pgp) encoded by the MDR1 gene leads to extrusion of anthracyclins, epipodophyllotoxins, Vinca alkaloids, coelenterazine and other cytostatic drugs.
The activity of the verapamil-sensitive P-glycoprotein (Pgp) encoded by the MDR1 gene leads to extrusion of anthracyclins, epipodophyllotoxins, Vinca alkaloids, coelenterazine and other cytostatic drugs.![]() BODIPY FL paclitaxel (P7500; Probes for Tubulin and Other Cytoskeletal Proteins—Section 11.2) is a substrate for Pgp-mediated transport that has been used as a probe for tumor spheroids.
BODIPY FL paclitaxel (P7500; Probes for Tubulin and Other Cytoskeletal Proteins—Section 11.2) is a substrate for Pgp-mediated transport that has been used as a probe for tumor spheroids.![]()
Many tumor cells do not express Pgp but export daunorubicin via a second, energy-dependent drug export mechanism.![]() A third, energy-dependent drug exporting mechanism is associated with a MDR-associated protein
A third, energy-dependent drug exporting mechanism is associated with a MDR-associated protein ![]() (MRP), which shows high selectivity toward glutathione S-conjugates and is inhibited by Vinca alkaloids and probenecid.
(MRP), which shows high selectivity toward glutathione S-conjugates and is inhibited by Vinca alkaloids and probenecid.![]() A fourth vanadate- and verapamil-resistant but probenecid-sensitive glutathione S-conjugate–exporting system in mouse and rat fibroblasts has also been reported.
A fourth vanadate- and verapamil-resistant but probenecid-sensitive glutathione S-conjugate–exporting system in mouse and rat fibroblasts has also been reported.![]() The probes commonly used to follow the transport of glutathione adducts are the same probes used to measure intracellular levels of glutathione (see below): monochlorobimane
The probes commonly used to follow the transport of glutathione adducts are the same probes used to measure intracellular levels of glutathione (see below): monochlorobimane ![]() (M1381MP) and CellTracker Green CMFDA
(M1381MP) and CellTracker Green CMFDA ![]() (C2925, C7025).
(C2925, C7025).
Obviously, the mechanisms of MDR are complex and, in some cases, have overlapping selectivity for the substrates.![]() We offer the Vybrant Multidrug Resistance Assay Kit, along with a variety of useful fluorescent probes for monitoring various aspects of the MDR phenotype.
We offer the Vybrant Multidrug Resistance Assay Kit, along with a variety of useful fluorescent probes for monitoring various aspects of the MDR phenotype.
MDR Assays Using Acetoxymethyl Esters
The discovery that fluorescent calcium indicators such as indo-1 AM and fluo-3 AM (I1203, F1241, F23915; Fluorescent Ca2+ Indicators Excited with Visible Light—Section 19.3) and other dyes such as calcein AM (C1430, C3099, C3100MP), are rapidly extruded from cells expressing Pgp ![]() presented a new class of highly sensitive probes for functional assays of the MDR1-encoded Pgp. Calcein AM—but not calcein—is an activator of Pgp in isolated membranes with a Kd ≤1 µM.
presented a new class of highly sensitive probes for functional assays of the MDR1-encoded Pgp. Calcein AM—but not calcein—is an activator of Pgp in isolated membranes with a Kd ≤1 µM.![]() Cells expressing the MDR1-encoded Pgp rapidly remove the nonfluorescent probe calcein AM, resulting in decreased accumulation of the highly fluorescent calcein in the cytoplasmic compartment.
Cells expressing the MDR1-encoded Pgp rapidly remove the nonfluorescent probe calcein AM, resulting in decreased accumulation of the highly fluorescent calcein in the cytoplasmic compartment.![]() Calcein AM is also a substrate for the MDR-associated protein (MRP), although in this case the hydrophilic free calcein anion is also exported.
Calcein AM is also a substrate for the MDR-associated protein (MRP), although in this case the hydrophilic free calcein anion is also exported.![]() Because calcein itself is not a substrate for the MDR1-encoded Pgp, MDR can be quantitatively assessed by measuring the net accumulation of intracellular fluorescence.
Because calcein itself is not a substrate for the MDR1-encoded Pgp, MDR can be quantitatively assessed by measuring the net accumulation of intracellular fluorescence.![]() Cellular depletion of glutathione does not affect the extrusion of calcein AM by the MDR1-encoded Pgp.
Cellular depletion of glutathione does not affect the extrusion of calcein AM by the MDR1-encoded Pgp.![]() Fluorescence of intracellular calcein can be distinguished from that of calcein that has leaked into the medium by adding Co2+ to totally quench the fluorescence of the extracellular dye.
Fluorescence of intracellular calcein can be distinguished from that of calcein that has leaked into the medium by adding Co2+ to totally quench the fluorescence of the extracellular dye.![]() The patented calcein AM assay for Pgp-related MDR is suitable for either flow cytometry
The patented calcein AM assay for Pgp-related MDR is suitable for either flow cytometry ![]() or fluorometry and is more rapid and significantly more sensitive than conventional assays based on doxorubicin accumulation.
or fluorometry and is more rapid and significantly more sensitive than conventional assays based on doxorubicin accumulation.![]() Reduced accumulation of calcein in MDR cells can also be observed in single cells by fluorescence microscopy.
Reduced accumulation of calcein in MDR cells can also be observed in single cells by fluorescence microscopy.![]()
Vybrant Multidrug Resistance Assay Kit
The Vybrant Multidrug Resistance Assay Kit (V13180), which is based on the fluorescence microplate–based method developed by Tiberghien and Loor,![]() provides a rapid and simple method for large-scale screening of MDR inhibitors. This assay utilizes the fluorogenic dye calcein AM as a substrate for efflux activity of Pgp. Upon hydrolysis by intracellular esterases, calcein is well retained in the cytosol and, unlike the hydrolysis product of other fluorescent Pgp substrates such as BCECF AM or fura-2 AM, its fluorescence is neither pH nor calcium dependent. MDR cells expressing high levels of Pgp rapidly extrude nonfluorescent calcein AM from the plasma membrane, reducing accumulation of fluorescent calcein in the cytosol
provides a rapid and simple method for large-scale screening of MDR inhibitors. This assay utilizes the fluorogenic dye calcein AM as a substrate for efflux activity of Pgp. Upon hydrolysis by intracellular esterases, calcein is well retained in the cytosol and, unlike the hydrolysis product of other fluorescent Pgp substrates such as BCECF AM or fura-2 AM, its fluorescence is neither pH nor calcium dependent. MDR cells expressing high levels of Pgp rapidly extrude nonfluorescent calcein AM from the plasma membrane, reducing accumulation of fluorescent calcein in the cytosol ![]() (Figure 15.6.5). The amount of Pgp activity is inversely proportional to the accumulation of intracellular calcein fluorescence. This assay is designed for use with fluorescence microplate readers and is particularly useful for rapid and sensitive screening of candidate Pgp inhibitors in MDR cell lines. The absorption/emission maxima of calcein (494/517 nm) are ideally suited for detection by instruments equipped with standard fluorescein filters. The Vybrant Multidrug Resistance Assay Kit (V13180) contains sufficient reagents to perform approximately 10,000 assays using a fluorescence microplate reader, including:
(Figure 15.6.5). The amount of Pgp activity is inversely proportional to the accumulation of intracellular calcein fluorescence. This assay is designed for use with fluorescence microplate readers and is particularly useful for rapid and sensitive screening of candidate Pgp inhibitors in MDR cell lines. The absorption/emission maxima of calcein (494/517 nm) are ideally suited for detection by instruments equipped with standard fluorescein filters. The Vybrant Multidrug Resistance Assay Kit (V13180) contains sufficient reagents to perform approximately 10,000 assays using a fluorescence microplate reader, including:
- Calcein AM
- Cyclosporin A, a competitive inhibitor of drug binding to Pgp
- Verapamil, a calcium channel blocker that noncompetitively inhibits Pgp activity
- Detailed protocols (Vybrant Multidrug Resistance Assay Kit)
 | Figure 15.6.5 Principle of the Vybrant Multidrug Resistance Assay Kit (V13180). In normal cells, nonfluorescent calcein AM readily diffuses across the cell membrane. Fluorescent calcein accumulates in the cytoplasm after cleavage of calcein AM by endogenous esterases. In MDR cells, overexpression of MDR transporter proteins increases expulsion of calcein AM from the cell membrane before enzymatic hydrolysis of its AM esters, thus reducing accumulation of intracellular calcein. |
MDR Assays Using Mitochondrial Probes
In the classical functional assay for MDR, doxorubicin efflux is measured.![]() The principal weakness of this assay is its low sensitivity, which has stimulated the search for other fluorochromes to monitor drug efflux. MDR cells overexpressing the Pgp have been identified using various mitochondrial probes (Probes for Mitochondria—Section 12.2), including rhodamine 123 (R302, R22420), acridine orange 10-nonyl bromide (nonyl acridine orange, A1372) and rhodamine 6G
The principal weakness of this assay is its low sensitivity, which has stimulated the search for other fluorochromes to monitor drug efflux. MDR cells overexpressing the Pgp have been identified using various mitochondrial probes (Probes for Mitochondria—Section 12.2), including rhodamine 123 (R302, R22420), acridine orange 10-nonyl bromide (nonyl acridine orange, A1372) and rhodamine 6G ![]() (R634). Furthermore, it has been reported that the fluorescence excitation spectrum of rhodamine 123 is different in drug-resistant and drug-sensitive cells.
(R634). Furthermore, it has been reported that the fluorescence excitation spectrum of rhodamine 123 is different in drug-resistant and drug-sensitive cells.![]()
The potential-sensitive carbocyanine dyes, including dipentyl-, dihexyl- and diheptyloxacarbocyanines (D272, D273, D378; Slow-Response Probes—Section 22.3), also have advantages over the common doxorubicin assay for MDR.![]() Not only are these carbocyanine dyes more fluorescent, permitting use of lower dye concentrations, but their fluorescence increases upon binding to cell membranes,
Not only are these carbocyanine dyes more fluorescent, permitting use of lower dye concentrations, but their fluorescence increases upon binding to cell membranes,![]() unlike the fluorescence of doxorubicin, which is substantially quenched inside cells.
unlike the fluorescence of doxorubicin, which is substantially quenched inside cells.![]() The ratiometric, potential-sensitive di-4-ANEPPS probe (D1199, Fast-Response Probes—Section 22.2) has been used to demonstrate that MDR cells have decreased electrical potentials.
The ratiometric, potential-sensitive di-4-ANEPPS probe (D1199, Fast-Response Probes—Section 22.2) has been used to demonstrate that MDR cells have decreased electrical potentials.![]()
MDR Assays Using Nucleic Acid Stains
MDR cells have also been identified by their decreased accumulation of nucleic acid–binding dyes such as Hoechst 33258 (H1398, H3569, H21491; Nucleic Acid Stains—Section 8.1), Hoechst 33342 (H1399, H3570, H21492; Nucleic Acid Stains—Section 8.1) and ethidium bromide ![]() (E1305, E3565; Nucleic Acid Stains—Section 8.1). SYTO 16 green-fluorescent nucleic acid stain (S7578, Nucleic Acid Stains—Section 8.1) was shown to be a useful substrate for detecting Pgp-mediated resistance by flow cytometry and in single cells by confocal laser-scanning microscopy.
(E1305, E3565; Nucleic Acid Stains—Section 8.1). SYTO 16 green-fluorescent nucleic acid stain (S7578, Nucleic Acid Stains—Section 8.1) was shown to be a useful substrate for detecting Pgp-mediated resistance by flow cytometry and in single cells by confocal laser-scanning microscopy.![]() Our four SYTO Fluorescent Nucleic Acid Stain Sampler Kits (S7572, S11340, S11350, S11360; Nucleic Acid Stains—Section 8.1) provide samples of SYTO dyes with fluorescence covering the entire visible spectrum; these dyes may be screened for their utility in detecting MDR cells.
Our four SYTO Fluorescent Nucleic Acid Stain Sampler Kits (S7572, S11340, S11350, S11360; Nucleic Acid Stains—Section 8.1) provide samples of SYTO dyes with fluorescence covering the entire visible spectrum; these dyes may be screened for their utility in detecting MDR cells.
BODIPY FL Verapamil and Dihydropyridine
The Ca2+-channel blocker verapamil is one of several molecules known to inhibit Pgp-mediated drug efflux.![]() We offer the green-fluorescent BODIPY FL verapamil probe (B7431) for the study of Pgp function and localization. Verapamil appears to inhibit drug efflux by acting as a substrate for Pgp, thereby overwhelming the transporter's capacity to expel other drugs. Our BODIPY FL conjugate of verapamil, with spectral properties similar to fluorescein, also serves as a substrate for Pgp. This fluorescent verapamil derivative preferentially accumulates in the lysosomes of normal, drug-sensitive NIH 3T3 cells but is rapidly transported out of MDR cells, as revealed by fluorescence microscopy.
We offer the green-fluorescent BODIPY FL verapamil probe (B7431) for the study of Pgp function and localization. Verapamil appears to inhibit drug efflux by acting as a substrate for Pgp, thereby overwhelming the transporter's capacity to expel other drugs. Our BODIPY FL conjugate of verapamil, with spectral properties similar to fluorescein, also serves as a substrate for Pgp. This fluorescent verapamil derivative preferentially accumulates in the lysosomes of normal, drug-sensitive NIH 3T3 cells but is rapidly transported out of MDR cells, as revealed by fluorescence microscopy.![]() The outward transport of BODIPY FL verapamil from MDR cells is inhibited by underivatized verapamil, as well as by excess vinblastine.
The outward transport of BODIPY FL verapamil from MDR cells is inhibited by underivatized verapamil, as well as by excess vinblastine.![]()
Like verapamil, dihydropyridines are known to inhibit drug efflux. Consequently, our fluorescent dihydropyridines ![]() labeled with either the green-fluorescent DM-BODIPY (D7443, Probes for Ion Channels and Carriers—Section 16.3) or the orange-fluorescent ST-BODIPY (S7445, Probes for Ion Channels and Carriers—Section 16.3) fluorophores may be useful MDR probes.
labeled with either the green-fluorescent DM-BODIPY (D7443, Probes for Ion Channels and Carriers—Section 16.3) or the orange-fluorescent ST-BODIPY (S7445, Probes for Ion Channels and Carriers—Section 16.3) fluorophores may be useful MDR probes.
BODIPY FL Vinblastine
The antimitotic agent vinblastine inhibits the Pgp-mediated efflux of a number of drugs and other probes from multidrug-resistant cells. Our BODIPY FL conjugate of vinblastine (V12390) is useful as a highly fluorescent vinblastine analog.![]() A biologically active coumarin dye–labeled vinblastine has previously been described.
A biologically active coumarin dye–labeled vinblastine has previously been described.![]()
BODIPY FL Prazosin and BODIPY FL Forskolin
Photoaffinity analogs of prazosin, an α1-adrenergic receptor antagonist,![]() and forskolin, an adenylate cyclase activator, have been shown to selectively photolabel isolated Pgp.
and forskolin, an adenylate cyclase activator, have been shown to selectively photolabel isolated Pgp.![]() Our green-fluorescent BODIPY FL prazosin (B7433) and BODIPY FL forskolin (B7469) are useful tools for probing MDR mechanisms.
Our green-fluorescent BODIPY FL prazosin (B7433) and BODIPY FL forskolin (B7469) are useful tools for probing MDR mechanisms.![]()
Methotrexate Resistance and Gene Amplification
Tumor cells often undergo gene amplification that leads to overexpression of dihydrofolate reductase (DHFR). This increased DHFR expression (Figure 15.6.6) confers enhanced tolerance to the cytotoxic effects of methotrexate.![]() For the study of antimetabolite resistance and spontaneous gene amplification, we offer fluorescent methotrexate conjugates. In addition to green-fluorescent fluorescein methotrexate (M1198MP), which has been used to visualize biochemical networks in living cells,
For the study of antimetabolite resistance and spontaneous gene amplification, we offer fluorescent methotrexate conjugates. In addition to green-fluorescent fluorescein methotrexate (M1198MP), which has been used to visualize biochemical networks in living cells,![]() we provide Alexa Fluor 488 methotrexate (M23271), which exhibits fluorescein-like spectral characteristics. The quantitative binding of fluorescein methotrexate (M1198MP) to dihydrofolate reductase (DHFR) enables researchers to isolate cells based on DHFR expression.
we provide Alexa Fluor 488 methotrexate (M23271), which exhibits fluorescein-like spectral characteristics. The quantitative binding of fluorescein methotrexate (M1198MP) to dihydrofolate reductase (DHFR) enables researchers to isolate cells based on DHFR expression.![]() Our fluorescein cadaverine adduct, which was originally described by Gapski and colleagues,
Our fluorescein cadaverine adduct, which was originally described by Gapski and colleagues,![]() appears to be equivalent to fluorescein lysine methotrexate in its applications.
appears to be equivalent to fluorescein lysine methotrexate in its applications.![]()
 | Figure 15.6.6 Analysis of dihydrofolate reductase (DHFR) content in CHO cells. A mixture of DHFR+ and DHFR- cells was stained with 1 µM fluorescein methotrexate (M1198MP) for 2 hours at 37°C. Following incubation with fluorescein methotrexate, cells were trypsinized, washed in phosphate-buffered saline and analyzed by flow cytometry using 488 nm excitation. Emission was collected at 525 nm. |
The tripeptide glutathione (γ-L-glutamyl-L-cysteinylglycine) is the most abundant (up to 10 mM in the cytoplasm and about 1 mM in blood ![]() ) and important nonprotein thiols in mammalian cells. Glutathione plays a central role in protecting cells of all organs, including the brain,
) and important nonprotein thiols in mammalian cells. Glutathione plays a central role in protecting cells of all organs, including the brain,![]() against damage produced by free radicals, oxidants and electrophiles. A distinct mechanism of MDR involves the overexpression of energy-dependent membrane pumps dedicated to removal of glutathione S-conjugates from the cytoplasm by a multidrug resistance–associated protein
against damage produced by free radicals, oxidants and electrophiles. A distinct mechanism of MDR involves the overexpression of energy-dependent membrane pumps dedicated to removal of glutathione S-conjugates from the cytoplasm by a multidrug resistance–associated protein ![]() (MRP). An increased rate of efflux of glutathione from Jurkat T lymphocytes during anti-FAS/APO-1 antibody–induced apoptosis has been reported.
(MRP). An increased rate of efflux of glutathione from Jurkat T lymphocytes during anti-FAS/APO-1 antibody–induced apoptosis has been reported.![]()
Several fluorescent reagents have been proposed for determining cellular levels of glutathione and glutathione S-transferase (GST), which catalyzes the formation of glutathione S-conjugates, but no probe is without drawbacks in quantitative studies of live cells. The high but variable levels of intracellular glutathione and the multitude of GST isozymes make kinetic measurements under saturating substrate conditions difficult or impossible.![]() Isozymes of GST vary both in abundance and activity, further complicating the analysis.
Isozymes of GST vary both in abundance and activity, further complicating the analysis.![]() Moreover, the fluorescent reagents designed to measure glutathione may react with intracellular thiols other than glutathione, including proteins in glutathione-depleted cells.
Moreover, the fluorescent reagents designed to measure glutathione may react with intracellular thiols other than glutathione, including proteins in glutathione-depleted cells.![]() Therefore, precautions must be taken in applying the reagents mentioned here to quantitate either glutathione or GST in cells. A useful strategy is to test a variety of glutathione-sensitive dyes—those requiring glutathione S-transferase activity, as well as GST-independent fluorophores—under controlled experimental conditions in which glutathione is depleted.
Therefore, precautions must be taken in applying the reagents mentioned here to quantitate either glutathione or GST in cells. A useful strategy is to test a variety of glutathione-sensitive dyes—those requiring glutathione S-transferase activity, as well as GST-independent fluorophores—under controlled experimental conditions in which glutathione is depleted.![]()
ThiolTracker Violet Glutathione Detection Reagent
The ThiolTracker Violet reagent (T10095, T10096) reacts with reduced thiols in intact cells and is up to 10-fold brighter than the bimanes traditionally used for glutathione (GSH) detection. Staining is achieved by applying ThiolTracker Violet dye to live cells in thiol-free buffer and then directly imaging labeled cells using excitation with either a 405 nm violet diode laser or conventional xenon or mercury arc lamps (excitation/emission maxima ~404/526 nm). Alternatively, because this cell-permeant stain survives formaldehyde fixation and detergent extraction, labeled cells can be subjected to immunochemical analysis prior to imaging.
Glutathione Determination with Monochlorobimane
Cell-permeant monochlorobimane (mBCl, M1381MP), which is essentially nonfluorescent until conjugated to thiols, has long been the preferred thiol-reactive probe for quantitating glutathione levels in cells and for measuring GST activity.![]() Because the blue-fluorescent glutathione adduct of monochlorobimane eventually accumulates in the nucleus, it is not a reliable indicator of the nuclear and cytoplasmic distribution of cellular glutathione.
Because the blue-fluorescent glutathione adduct of monochlorobimane eventually accumulates in the nucleus, it is not a reliable indicator of the nuclear and cytoplasmic distribution of cellular glutathione.![]() Tissue glutathione levels can also be measured fluorometrically by adding both monochlorobimane and glutathione S-transferase to tissue homogenates.
Tissue glutathione levels can also be measured fluorometrically by adding both monochlorobimane and glutathione S-transferase to tissue homogenates.![]()
Monochlorobimane is reported to react more selectively with glutathione in whole cells than does monobromobimane (M1378, M20381; Thiol-Reactive Probes Excited with Ultraviolet Light—Section 2.3) and has proven useful for assaying drug resistance by flow cytometry ![]() and by fluorescence microscopy.
and by fluorescence microscopy.![]() Moreover, HPLC analysis has shown that glutathione is the only low molecular weight thiol in hepatocytes that reacts with monochlorobimane.
Moreover, HPLC analysis has shown that glutathione is the only low molecular weight thiol in hepatocytes that reacts with monochlorobimane.![]() Results from glutathione determination with monochlorobimane have been shown to match those from an independent glutathione-specific assay using glutathione reductase.
Results from glutathione determination with monochlorobimane have been shown to match those from an independent glutathione-specific assay using glutathione reductase.![]() However, although monochlorobimane was shown to be highly selective for glutathione in rodent cells, it was reported to inadequately label glutathione in human cells because of its low affinity for human glutathione S-transferases.
However, although monochlorobimane was shown to be highly selective for glutathione in rodent cells, it was reported to inadequately label glutathione in human cells because of its low affinity for human glutathione S-transferases.![]() The reducing agent tris-(2-carboxyethyl)phosphine (TCEP, T2556; Introduction to Thiol Modification and Detection—Section 2.1) has been used in place of dithiothreitol (DTT, D1532; Introduction to Thiol Modification and Detection—Section 2.1) in a simplified monobromobimane-based assay for glutathione
The reducing agent tris-(2-carboxyethyl)phosphine (TCEP, T2556; Introduction to Thiol Modification and Detection—Section 2.1) has been used in place of dithiothreitol (DTT, D1532; Introduction to Thiol Modification and Detection—Section 2.1) in a simplified monobromobimane-based assay for glutathione ![]() and thus may also prove useful for monochlorobimane-based assays. In this monobromobimane-based glutathione assay, an extraction step is reportedly necessary to remove a fluorescent, reductive-dehalogenation side product of TCEP and monobromobimane.
and thus may also prove useful for monochlorobimane-based assays. In this monobromobimane-based glutathione assay, an extraction step is reportedly necessary to remove a fluorescent, reductive-dehalogenation side product of TCEP and monobromobimane.![]() Probenecid (P36400, Chelators, Calibration Buffers, Ionophores and Cell-Loading Reagents—Section 19.8) inhibits the ATP-dependent organic anion pump and blocks the loss of the fluorescent bimane–glutathione adduct from rat fibroblasts.
Probenecid (P36400, Chelators, Calibration Buffers, Ionophores and Cell-Loading Reagents—Section 19.8) inhibits the ATP-dependent organic anion pump and blocks the loss of the fluorescent bimane–glutathione adduct from rat fibroblasts.![]() Monochlorobimane has also been employed to sort cells based on their expression of recombinant GST.
Monochlorobimane has also been employed to sort cells based on their expression of recombinant GST.![]()
Glutathione Determination with Visible Light–Excitable Thiol-Reactive Probes
CellTracker Green CMFDA (5-chloromethylfluorescein diacetate, C2925, C7025) is a useful alternative to the UV light–excitable monochlorobimane for determining levels of intracellular glutathione.![]() CellTracker Green CMFDA can be excited by the argon-ion laser, and is compatible with flow cytometry and confocal laser-scanning microscopy applications. CellTracker Green CMFDA's enzymatic product has a much higher absorbance and fluorescence quantum yield than that of monochlorobimane. In conjunction with Hoechst 33342 (H1399, H3570, H21492; Nucleic Acid Stains—Section 8.1), CellTracker Green CMFDA has also been shown to be effective for analyzing intracellular thiol levels as a function of cell cycle using flow cytometry,
CellTracker Green CMFDA can be excited by the argon-ion laser, and is compatible with flow cytometry and confocal laser-scanning microscopy applications. CellTracker Green CMFDA's enzymatic product has a much higher absorbance and fluorescence quantum yield than that of monochlorobimane. In conjunction with Hoechst 33342 (H1399, H3570, H21492; Nucleic Acid Stains—Section 8.1), CellTracker Green CMFDA has also been shown to be effective for analyzing intracellular thiol levels as a function of cell cycle using flow cytometry,![]() for following transport of the glutathione adduct to secretory vesicles in multidrug-resistant cells
for following transport of the glutathione adduct to secretory vesicles in multidrug-resistant cells ![]() and for detecting apoptotic cells, which have reduced levels of intracellular reduced glutathione.
and for detecting apoptotic cells, which have reduced levels of intracellular reduced glutathione.![]() Selectivity of CellTracker Green CMFDA for glutathione (versus thiolated proteins) was shown by the isolation of >95% of the intracellular fluorescent products as a mixture of the glutathione adduct and the unconjugated hydrolysis product, chloromethylfluorescein.
Selectivity of CellTracker Green CMFDA for glutathione (versus thiolated proteins) was shown by the isolation of >95% of the intracellular fluorescent products as a mixture of the glutathione adduct and the unconjugated hydrolysis product, chloromethylfluorescein.![]() However, in these experiments, the high fluorescence of unconjugated chloromethylfluorescein resulted in significantly increased background levels. Because glutathione-depleting chemicals may also cause cell death, it has been recommended that calcein AM be used to independently assess cell viability in assays that use CellTracker Green CMFDA to measure changes in the level of intracellular glutathione.
However, in these experiments, the high fluorescence of unconjugated chloromethylfluorescein resulted in significantly increased background levels. Because glutathione-depleting chemicals may also cause cell death, it has been recommended that calcein AM be used to independently assess cell viability in assays that use CellTracker Green CMFDA to measure changes in the level of intracellular glutathione.![]()
Like CMFDA, chloromethyl SNARF-1 acetate (C6826) forms adducts with intracellular thiols that are well retained by viable cells. The glutathione adduct of chloromethyl SNARF-1 can be excited by the 488 nm spectral line of the argon-ion laser yet emits beyond 630 nm, which may prove advantageous in multicolor applications or when assaying autofluorescent samples. A number of our other CellTracker (Membrane-Permeant Reactive Tracers—Section 14.2) and MitoTracker (Probes for Mitochondria—Section 12.2) probes have thiol-reactive chloromethyl moieties and may be similarly useful for glutathione determination. All of these probes form glutathione S-conjugates that are likely to be transported from the cytoplasm by an MDR-associated protein ![]() (MRP).
(MRP).
Glutathiolation Detection with BioGEE
Biotinylated glutathione ethyl ester (BioGEE, G36000) is a cell-permeant, biotinylated glutathione analog for detecting glutathiolation. Under conditions of oxidative stress, cells may transiently incorporate glutathione into proteins. Stressed cells incubated with BioGEE will also incorporate this biotinylated glutathione derivative into proteins, facilitating the identification of oxidation-sensitive proteins.![]() Once these cells are fixed and permeabilized, glutathiolation levels can be detected with a fluorescent streptavidin conjugate (Avidin, Streptavidin, NeutrAvidin and CaptAvidin Biotin-Binding Proteins and Affinity Matrices—Section 7.6, Molecular Probes avidin, streptavidin, NeutrAvidin and CaptAvidin conjugates—Table 7.9) using either flow cytometry or fluorescence microscopy. Proteins glutathiolated with BioGEE can also be extracted and analyzed by mass spectrometry or by western blotting methods in conjunction with fluorophore- or enzyme-labeled streptavidin conjugates.
Once these cells are fixed and permeabilized, glutathiolation levels can be detected with a fluorescent streptavidin conjugate (Avidin, Streptavidin, NeutrAvidin and CaptAvidin Biotin-Binding Proteins and Affinity Matrices—Section 7.6, Molecular Probes avidin, streptavidin, NeutrAvidin and CaptAvidin conjugates—Table 7.9) using either flow cytometry or fluorescence microscopy. Proteins glutathiolated with BioGEE can also be extracted and analyzed by mass spectrometry or by western blotting methods in conjunction with fluorophore- or enzyme-labeled streptavidin conjugates.
Glutathione Determination With o-Phthaldialdehyde and Naphthalene-2,3-dicarboxaldehyde
The reagent o-phthaldialdehyde (OPA, P2331MP) reacts with both the thiol and the amine functions of glutathione, yielding a cyclic derivative that is fluorescent. The spectra of the glutathione adduct of OPA (excitation/emission maxima of 350/420 nm) are shifted from those of its protein adducts.![]() This effect has occasionally been used to estimate glutathione levels in cells.
This effect has occasionally been used to estimate glutathione levels in cells.![]() OPA has also been used as a derivatization reagent for the chromatographic determination of glutathione in cells, blood and tissues.
OPA has also been used as a derivatization reagent for the chromatographic determination of glutathione in cells, blood and tissues.![]()
The membrane-permeant naphthalene-2,3-dicarboxaldehyde (NDA, N1138) has been used to determine glutathione levels in single cells. Cells were treated with the NDA reagent and then analyzed by capillary electrophoresis;![]() glutathione labeling was reported to be complete within two minutes. The glutathione adduct of NDA can be excited by the 458 nm spectral line of the argon-ion laser.
glutathione labeling was reported to be complete within two minutes. The glutathione adduct of NDA can be excited by the 458 nm spectral line of the argon-ion laser.
| Cat # | MW | Storage | Soluble | Abs | EC | Em | Solvent | Notes |
|---|---|---|---|---|---|---|---|---|
| B1150 | ~615 | F,D | DMSO | <300 | none | 1, 2 | ||
| B1170 | ~615 | F,D | DMSO | <300 | none | 1, 2 | ||
| B3051 | ~615 | F,D | DMSO | <300 | none | 1, 2, 3 | ||
| B7431 | 769.18 | F,D,L | DMSO, EtOH | 504 | 74,000 | 511 | MeOH | |
| B7433 | 563.41 | F,D,L | DMSO, EtOH | 504 | 77,000 | 511 | MeOH | |
| B7469 | 784.70 | F,D,L | DMSO | 504 | 79,000 | 511 | MeOH | |
| C1430 | 994.87 | F,D | DMSO | <300 | none | 4 | ||
| C2925 | 464.86 | F,D | DMSO | <300 | none | 5 | ||
| C3099 | 994.87 | F,D | DMSO | <300 | none | 3, 4 | ||
| C3100MP | 994.87 | F,D | DMSO | <300 | none | 4 | ||
| C6826 | 499.95 | F,D | DMSO | <350 | none | 6 | ||
| C7025 | 464.86 | F,D | DMSO | <300 | none | 5 | ||
| F1314 | 1213.41 | F,L | pH >6, DMF | 494 | 72,000 | 517 | pH 9 | |
| G36000 | 561.67 | F,D | DMSO | <300 | none | |||
| M1198MP | 979.08 | F,L | pH >6, DMF | 496 | 67,000 | 516 | pH 9 | |
| M1381MP | 226.66 | F,L | DMSO | 380 | 6000 | see Notes | MeOH | 7 |
| M23271 | 1055.06 | F,D,L | DMSO | 494 | 78,000 | 518 | pH 7 | |
| N1138 | 184.19 | L | DMF, MeCN | 419 | 9400 | 493 | see Notes | 8 |
| P2331MP | 134.13 | L | EtOH | 334 | 5700 | 455 | pH 9 | 9 |
| V12390 | 1043.02 | F,D,L | DMSO, DMF | 503 | 83,000 | 510 | MeOH | |
| ||||||||
For Research Use Only. Not for use in diagnostic procedures.