Search
Overview of Post-Translational Modifications (PTMs)
Protein post-translational modifications (PTMs) increase the functional diversity of the proteome by the covalent addition of functional groups or proteins, proteolytic cleavage of regulatory subunits, or degradation of entire proteins. These modifications include phosphorylation, glycosylation, ubiquitination, nitrosylation, methylation, acetylation, lipidation and proteolysis and influence almost all aspects of normal cell biology and pathogenesis. Therefore, identifying and understanding PTMs is critical in the study of cell biology and disease treatment and prevention.
Introduction
Within the last few decades, scientists have discovered that the human proteome is vastly more complex than the human genome. While it is estimated that the human genome comprises between 20,000 and 25,000 genes, the total number of proteins in the human proteome is estimated at over 1 million. These estimations demonstrate that single genes encode multiple proteins. Genomic recombination, transcription initiation at alternative promoters, differential transcription termination, and alternative splicing of the transcript are mechanisms that generate different mRNA transcripts from a single gene.
The increase in complexity from the level of the genome to the proteome is further facilitated by protein post-translational modifications (PTMs). PTMs are chemical modifications that play a key role in functional proteomic because they regulate activity, localization, and interaction with other cellular molecules such as proteins, nucleic acids, lipids and cofactors.
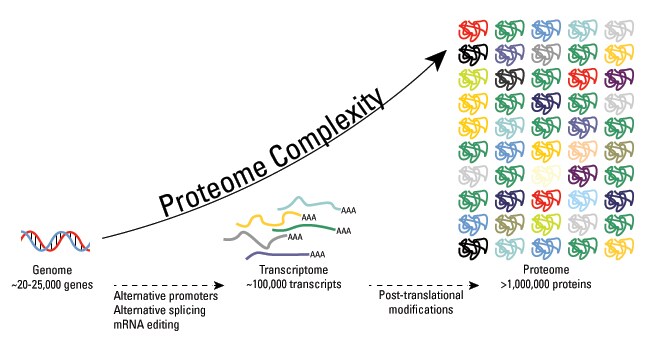
Post-translational modifications are key mechanisms to increase proteomic diversity. While the genome comprises 20,000 to 25,000 genes, the proteome is estimated to encompass over 1 million proteins. Changes at the transcriptional and mRNA levels increase the size of the transcriptome relative to the genome, and the myriad of different post-translational modifications exponentially increases the complexity of the proteome relative to both the transcriptome and genome.
Additionally, the human proteome is dynamic and changes in response to a legion of stimuli, and post-translational modifications are commonly employed to regulate cellular activity. PTMs occur at distinct amino acid side chains or peptide linkages, and they are most often mediated by enzymatic activity. Indeed, it is estimated that 5% of the proteome comprises enzymes that perform more than 200 types of post-translational modifications. These enzymes include kinases, phosphatases, transferases and ligases, which add or remove functional groups, proteins, lipids or sugars to or from amino acid side chains; and proteases, which cleave peptide bonds to remove specific sequences or regulatory subunits. Many proteins can also modify themselves using autocatalytic domains, such as autokinase and autoprotolytic domains.
Post-translational modification can occur at any step in the "life cycle" of a protein. For example, many proteins are modified shortly after translation is completed to mediate proper protein folding or stability or to direct the nascent protein to distinct cellular compartments (e.g., nucleus, membrane). Other modifications occur after folding and localization are completed to activate or inactivate catalytic activity or to otherwise influence the biological activity of the protein. Proteins are also covalently linked to tags that target a protein for degradation. Besides single modifications, proteins are often modified through a combination of post-translational cleavage and the addition of functional groups through a step-wise mechanism of protein maturation or activation.
Protein PTMs can also be reversible depending on the nature of the modification. For example, kinases phosphorylate proteins at specific amino acid side chains, which is a common method of catalytic activation or inactivation. Conversely, phosphatases hydrolyze the phosphate group to remove it from the protein and reverse the biological activity. Proteolytic cleavage of peptide bonds is a thermodynamically favorable reaction and therefore permanently removes peptide sequences or regulatory domains.
Consequently, the analysis of proteins and their post-translational modifications is particularly important for the study of heart disease, cancer, neurodegenerative diseases and diabetes. The characterization of PTMs, although challenging, provides invaluable insight into the cellular functions underlying etiological processes. Technically, the main challenges to studying post-translationally modified proteins are the development of specific detection and purification methods. Fortunately, these technical obstacles are being overcome with a variety of new and refined proteomics technologies.
Protein Expression Handbook
This 118-page handbook provides comprehensive information about protein expression and will help you choose the right expression system and purification technologies for your specific application and needs. Get tips and tricks when starting an experiment, and find answers to everyday problems related to protein expression.

Post-translational modifications (PTMs)
As noted above, the large number of different PTMs precludes a thorough review of all possible protein modifications. Therefore, this overview only touches on a small number of the most common types of PTMs studied in protein research today. Furthermore, greater focus is placed on phosphorylation, glycosylation and ubiquitination, and therefore these PTMs are described in greater detail on pages dedicated to the respective PTM.
Phosphorylation
Reversible protein phosphorylation, principally on serine, threonine or tyrosine residues, is one of the most important and well-studied post-translational modifications. Phosphorylation plays critical roles in the regulation of many cellular processes, including cell cycle, growth, apoptosis and signal transduction pathways. In the following example, western blot analysis was used to evaluate phosphoprotein specificity in lysates obtained from serum-starved HeLa and NIH 3T3 cancer cell lines stimulated with epidermal growth factor (EGF) and platelet derived growth factor (PDGF), respectively.
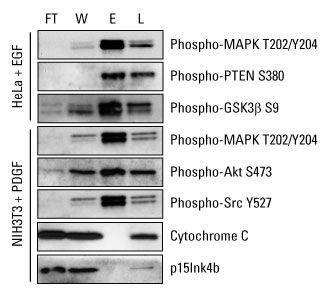
Highly pure phosphoprotein enrichment from complex biological samples. Western blot analysis was performed with the Thermo Scientific Pierce Phosphoprotein Enrichment Kit, and cell lysates were prepared according to the kit instructions to enrich for phosphoproteins. Protein detection was achieved using phospho-specific antibodies that recognize key regulatory proteins involved in growth factor signaling. Cytochrome C (pI 9.6) and p15Ink4b (pI 5.5) served as negative controls for nonspecific binding of non-phosphorylated proteins. FT = flow-through fraction, W = pooled wash fractions, E = pooled elution fractions and L = non-enriched total cell extract.
Learn more
- Phosphorylation
- Phosphoproteomics
- Application Note: Phosphoprotein Enrichment from Cell and Tissue Samples
- Protein Expression Support Center
Glycosylation
Protein glycosylation is acknowledged as one of the major post-translational modifications, with significant effects on protein folding, conformation, distribution, stability and activity. Glycosylation encompasses a diverse selection of sugar-moiety additions to proteins that ranges from simple monosaccharide modifications of nuclear transcription factors to highly complex branched polysaccharide changes of cell surface receptors. Carbohydrates in the form of asparagine-linked (N-linked) or serine/threonine-linked (O-linked) oligosaccharides are major structural components of many cell surface and secreted proteins.
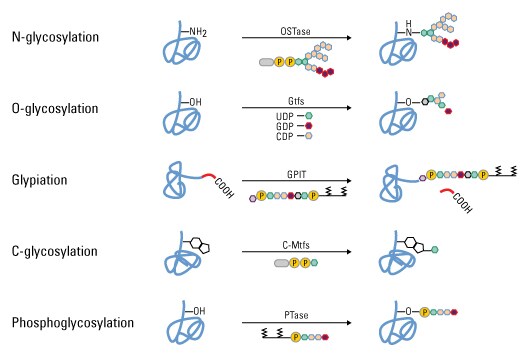
Types of glycosylation. Glycopeptide bonds can be categorized into specific groups based on the nature of the sugar–peptide bond and the oligosaccharide attached, including N-, O- and C-linked glycosylation, glypiation and phosphoglycosylation.
Ubiquitination
Ubiquitin is an 8-kDa polypeptide consisting of 76 amino acids that is appended to the ε-NH2 of lysine in target proteins via the C-terminal glycine of ubiquitin. Following an initial monoubiquitination event, the formation of a ubiquitin polymer may occur, and polyubiquitinated proteins are then recognized by the 26S proteasome that catalyzes the degradation of the ubiquitinated protein and the recycling of ubiquitin. The following experiment provides an example of methods used to detect ubiquitinated proteins.
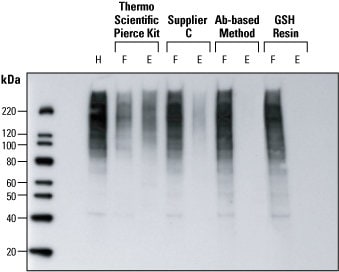
Detection of ubiquitin in HeLa cell lysates. Western blot analysis was performed to compare four methods for detecting ubiquitin protein in HeLa cell lysates. After epoxomicin-treatment, HeLa cells lysates (150 µg) were processed by four different methods. The resulting flow-through (F) and elution (E) fractions were volume-normalized to the original unprocessed lysate (H) and identical volumes electrophoresed for western blot detection. Compared to Supplier C’s kit and an antibody-based method, the Thermo Scientific Pierce Ubiquitin Enrichment Kit yielded more ubiquitinated protein in the elution fraction (and less protein in the flow-through fraction), indicating significantly better enrichment of ubiquitinated proteins. GSH Resin is a negative control for comparison.
S-nitrosylation
Nitric oxide (NO) is produced by three isoforms of nitric oxide synthase (NOS), and it is a chemical messenger that reacts with free cysteine residues to form S-nitrothiols (SNOs). S-nitrosylation is a critical PTM used by cells to stabilize proteins, regulate gene expression and provide NO donors, and the generation, localization, activation and catabolism of SNOs are tightly regulated.
S-nitrosylation is a reversible reaction, and SNOs have a short half-life in the cytoplasm because of the host of reducing enzymes, including glutathione (GSH) and thioredoxin, that denitrosylate proteins. Therefore, SNOs are often stored in membranes, vesicles, the interstitial space and lipophilic protein folds to protect them from denitrosylation. For example, caspases, which mediate apoptosis, are stored in the mitochondrial intermembrane space as SNOs. In response to extra- or intracellular cues, the caspases are released into the cytoplasm, and the highly reducing environment rapidly denitrosylates the proteins, resulting in caspase activation and the induction of apoptosis.
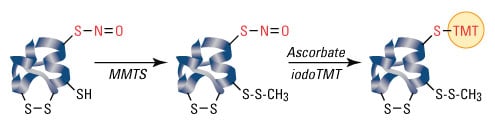
S-nitrosylation is not a random event, and only specific cysteine residues are S-nitrosylated. Because proteins may contain multiple cysteines and due to the labile nature of SNOs, S-nitrosylated cysteines can be difficult to detect and distinguish from non-S-nitrosylated amino acids. The biotin switch assay, developed by Jaffrey et al., is a common method of detecting SNOs, and the steps of the assay are listed below:
- All free cysteines are blocked.
- All remaining cysteines (presumably only those that are denitrosylated) are denitrosylated.
- The now-free thiol groups are then biotinylated.
- Biotinylated proteins are detected by SDS-PAGE and western blot analysis or mass spectrometry.
Learn more
- Biotinylation
- Overview of Protein Electrophoresis
- Overview of Western Blotting
- Application Note: Specific Labeling, Enrichment and Quantitation of S-nitrosylated Peptides Using iodoTMT Reagents
Methylation
The transfer of one-carbon methyl groups to nitrogen or oxygen (N- and O-methylation, respectively) to amino acid side chains increases the hydrophobicity of the protein and can neutralize a negative amino acid charge when bound to carboxylic acids. Methylation is mediated by methyltransferases, and S-adenosyl methionine (SAM) is the primary methyl group donor.
Methylation occurs so often that SAM has been suggested to be the most used substrate in enzymatic reactions after ATP. Additionally, while N-methylation is irreversible, O-methylation is potentially reversible. Methylation is a well-known mechanism of epigenetic regulation, as histone methylation and demethylation influences the availability of DNA for transcription. Amino acid residues can be conjugated to a single methyl group or multiple methyl groups to increase the effects of modification.
The figure below provides an illustration of PMTs associated with nucleosome core particles.
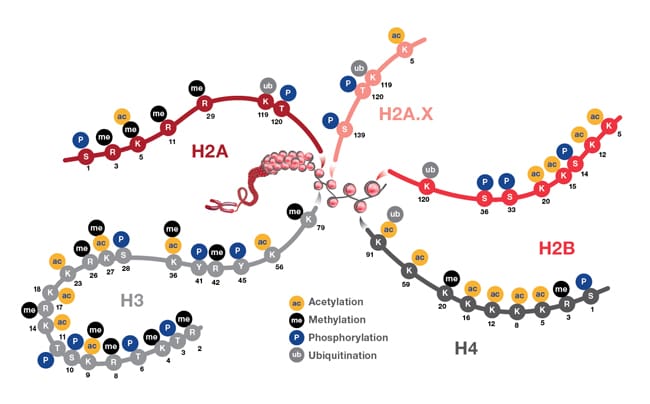
Representation showing post-translational modifications associated with histone particles. Nucleosomes are represented by red spheres wrapped by DNA (shown in gray). Also depicted are the positions of PTMs located on the histone proteins H2A (and H2A.X), H2B, H3, and H4. These PTMs impact gene expression by altering chromatin structure and recruiting histone modifiers. PTM events mediate diverse biological functions such as transcriptional activation and inactivation, chromosome packaging, and DNA damage and repair processes.
N-acetylation
N-acetylation, or the transfer of an acetyl group to nitrogen, occurs in almost all eukaryotic proteins through both irreversible and reversible mechanisms. N-terminal acetylation requires the cleavage of the N-terminal methionine by methionine aminopeptidase (MAP) before replacing the amino acid with an acetyl group from acetyl-CoA by N-acetyltransferase (NAT) enzymes. This type of acetylation is co-translational, in that N-terminus is acetylated on growing polypeptide chains that are still attached to the ribosome. While 80 to 90% of eukaryotic proteins are acetylated in this manner, the exact biological significance is still unclear.
Acetylation at the ε-NH2 of lysine (termed lysine acetylation) on histone N-termini is a common method of regulating gene transcription. Histone acetylation is a reversible event that reduces chromosomal condensation to promote transcription, and the acetylation of these lysine residues is regulated by transcription factors that contain histone acetyltransferase (HAT) activity. While transcription factors with HAT activity act as transcription co-activators, histone deacetylase (HDAC) enzymes are co-repressors that reverse the effects of acetylation by reducing the level of lysine acetylation and increasing chromosomal condensation.
Sirtuins (silent information regulator) are a group of NAD-dependent deacetylases that target histones. As their name implies, they maintain gene silencing by hypoacetylating histones and have been reported to aid in maintaining genomic stability.
While acetylation was first detected in histones, cytoplasmic proteins have been reported to also be acetylated, and therefore acetylation seems to play a greater role in cell biology than simply transcriptional regulation. Furthermore, crosstalk between acetylation and other post-translational modifications, including phosphorylation, ubiquitination and methylation, can modify the biological function of the acetylated protein.
Protein acetylation can be detected by chromatin immunoprecipitation (ChIP) using acetyllysine-specific antibodies or by mass spectrometry, where an increase in histone by 42 mass units represents a single acetylation.
Lipidation
Lipidation is a method to target proteins to membranes in organelles (endoplasmic reticulum [ER], Golgi apparatus, mitochondria), vesicles (endosomes, lysosomes) and the plasma membrane. The four types of lipidation are:
- C-terminal glycosyl phosphatidylinositol (GPI) anchor
- N-terminal myristoylation
- S-myristoylation
- S-prenylation
Each type of modification gives proteins distinct membrane affinities, although all types of lipidation increase the hydrophobicity of a protein and thus its affinity for membranes. The different types of lipidation are also not mutually exclusive, in that two or more lipids can be attached to a given protein.
GPI anchors tether cell surface proteins to the plasma membrane. These hydrophobic moieties are prepared in the ER, where they are then added to the nascent protein en bloc. GPI-anchored proteins are often localized to cholesterol- and sphingolipid-rich lipid rafts, which act as signaling platforms on the plasma membrane. This type of modification is reversible, as the GPI anchor can be released from the protein by phosphoinositol-specific phospholipase C. Indeed, this lipase is used in the detection of GPI-anchored proteins to release GPI-anchored proteins from membranes for gel separation and analysis by mass spectrometry.
N-myristoylation is a method to give proteins a hydrophobic handle for membrane localization. The myristoyl group is a 14-carbon saturated fatty acid (C14), which gives the protein sufficient hydrophobicity and affinity for membranes, but not enough to permanently anchor the protein in the membrane. N-myristoylation can therefore act as a conformational localization switch in which protein conformational changes influence the availability of the handle for membrane attachment. Because of this conditional localization, signal proteins that selectively localize to membrane, such as Src-family kinases, are N-myristoylated.
N-myristoylation is facilitated specifically by N-myristoyltransferase (NMT) and uses myristoyl-CoA as the substrate to attach the myristoyl group to the N-terminal glycine. Because methionine is the N-terminal amino acid of all eukaryotic proteins, this PTM requires methionine cleavage by the above-mentioned MAP prior to addition of the myristoyl group; this represents one example of multiple PTMs on a single protein.
S-palmitoylation adds a C16 palmitoyl group from palmitoyl-CoA to the thiolate side chain of cysteine residues via palmitoyl acyltransferases (PATs). Because of the longer hydrophobic group, this anchor can permanently anchor the protein to the membrane. This localization can be reversed, though, by thioesterases that break the link between the protein and the anchor; thus, S-palmitoylation is used as an on/off switch to regulate membrane localization. S-palmitoylation is often used to strengthen other types of lipidation, such as myristoylation or farnesylation (see below). S-palmitoylated proteins also selectively concentrate at lipid rafts.
S-prenylation covalently adds a farnesyl (C15) or geranylgeranyl (C20) group to specific cysteine residues within five amino acids from the C-terminus via farnesyl transferase (FT) or geranylgeranyl transferases (GGT I and II). Unlike S-palmitoylation, S-prenylation is hydrolytically stable. Approximately 2% of all proteins are prenylated, including all members of the Ras superfamily. This group of molecular switches is farnesylated, geranylgeranylated or a combination of both. Additionally, these proteins have specific 4-amino acid motifs at the C-terminus that determine the type of prenylation at single or dual cysteines. Prenylation occurs in the ER and is often part of a stepwise process of PTMs that is followed by proteolytic cleavage by Rce1 and methylation by isoprenyl cysteine methyltransferase (ICMT).
Select products
Proteolysis
Peptide bonds are indefinitely stable under physiological conditions, and therefore cells require some mechanism to break these bonds. Proteases comprise a family of enzymes that cleave the peptide bonds of proteins and are critical in antigen processing, apoptosis, surface protein shedding and cell signaling.
The family of over 11,000 proteases varies in substrate specificity, mechanism of peptide cleavage, location in the cell and the length of activity. While this variation suggests a wide array of functionalities, proteases can generally be separated into groups based on the type of proteolysis. Degradative proteolysis is critical to remove unassembled protein subunits and misfolded proteins and to maintain protein concentrations at homeostatic concentrations by reducing a given protein to the level of small peptides and single amino acids. Proteases also play a biosynthetic role in cell biology that includes cleaving signal peptides from nascent proteins and activating zymogens, which are inactive enzyme precursors that require cleavage at specific sites for enzyme function. In this respect, proteases act as molecular switches to regulate enzyme activity.
Proteolysis is a thermodynamically favorable and irreversible reaction. Therefore, protease activity is tightly regulated to avoid uncontrolled proteolysis through temporal and/or spatial control mechanisms including regulation by cleavage in cis or trans and compartmentalization (e.g., proteasomes, lysosomes).
The diverse family of proteases can be classified by the site of action, such as aminopeptidases and carboxypeptidase, which cleave at the amino or carboxy terminus of a protein, respectively. Another type of classification is based on the active site groups of a given protease that are involved in proteolysis. Based on this classification strategy, greater than 90% of known proteases fall into one of four categories as follows:
- Serine proteases
- Cysteine proteases
- Aspartic acid proteases
- Zinc metalloproteases
The following representative example demonstrates the performance of a commercially available protease assay.
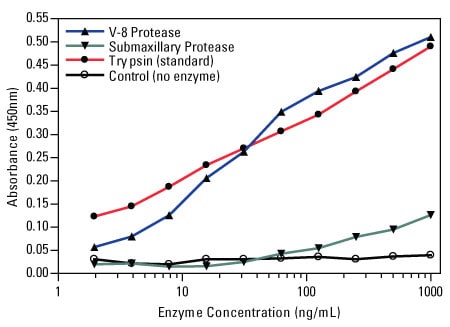
Colorimetric protease assay response curves. The Thermo Scientific Pierce Colorimetric Protease Assay Kit was used to measure the activity of V-8 protease and submaxillary protease for digestion of casein substrate by comparison to the supplied trypsin standard.
Recommended reading
- International Human Genome Sequencing Consortium (2004) Finishing the euchromatic sequence of the human genome. Nature 431:931–45.
- Jensen ON (2004) Modification-specific proteomics: Characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol 8:33–41.
- Ayoubi TA, Van De Ven WJ (1996) Regulation of gene expression by alternative promoters. FASEB J 10:453–60.
- Walsh C (2006) Posttranslational modification of proteins: Expanding nature's inventory. Englewood (CO): Roberts and Co. Publishers. xxi, p 490.
- Gaston BM et al. (2003) S-nitrosylation signaling in cell biology. Mol Interv 3:253–63.
- Jaffrey SR, Snyder SH (2001) The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 86:pl1.
- Han P, Chen C (2008) Detergent-free biotin switch combined with liquid chromatography/tandem mass spectrometry in the analysis of S-nitrosylated proteins. Rapid Commun Mass Spectrom 22:1137–45.
- Imai S et al. (2000) Transcriptional silencing and longevity protein SIR2 is an NAD-dependent histone deacetylase. Nature 403:795–800.
- Glozak MA et al. (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23.
- Yang XJ, Seto E (2008) Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol Cell 31:449–61.
For Research Use Only. Not for use in diagnostic procedures.